Heart Disease
If you don’t want to read all the details, scientific evidence, studies, etc., and just to get to the point of it all, this first section in bold is for you:
The conventional story says heart disease occurs because cholesterol clogs arteries, so the solution has been to lower cholesterol and replace saturated fat with vegetable oils. I believe the evidence points somewhere else. The deeper problem is polyunsaturated fat (PUFA) oxidation. Fragile polyunsaturated fats from many vegetable oils can become part of LDL particles, oxidize inside the artery wall, trigger inflammation, attract immune cells, build plaque, weaken the plaque cap, and increase the chance of rupture and clotting.
Below, the post will explore the concept of how mainstream medicine often confuses a repair signal with the original cause. Cholesterol is treated as the villain because it is found in damaged arteries, but that logic is like blaming garbage men for garbage appearing at the curb or firefighters appearing every time there’s a fire. Garbage men arrive because garbage is already there, and firefighters arrive because a fire has already started. In the same way, cholesterol may appear in injured or inflamed tissue because the body is using it for transport, protection, repair, and cleanup—not necessarily because cholesterol caused the damage in the first place.
The next question is why cholesterol gets high in the first place, and the answer is simple: high cholesterol is usually not a sign that the body has too much cholesterol, but that it is not using cholesterol well. Cholesterol is supposed to be converted into protective hormones and bile acids, but that conversion depends heavily on thyroid-driven metabolism. When thyroid function and energy production are low, cholesterol can back up in the blood instead of being used as designed. I believe that restoring adequate thyroid hormone action is the most effective way to bring cholesterol levels down, while sugar, protein, vitamin A, minerals, and low polyunsaturated-fat intake help support that process. The rest of this paper will explain that mechanism in more detail.
The practical takeaway in this is to reduce heavy vegetable-oil intake while supporting antioxidant defense, energy production, and tissue repair: vitamin E, vitamin C, glycine/collagen, adequate protein, copper, manganese, B vitamins, magnesium, potassium, selenium, zinc, and fat-soluble vitamins A, D, and K2. Thyroid function, digestion, and stable energy metabolism also belong in the same picture.
For those who want the full in-depth picture:
There are two broad ways the heart can be the cause of death: either the disease of the blood vessels that feed the heart or failure of the heart muscle itself. Both involve the same organ, but are not the same. The first half of this article will focus on the congestve heatt failure side of things: plaque, oxidation, narrowing, rupture, and clotting. The second half will focus on the heart-muscle side: energy failure, weak contraction, poor relaxation, and failed repair.
Heart disease is the leading cause of death in the United States. According to CDC/NCHS FastStats, it killed 683,491 Americans in 2024, more than cancer, accidents, stroke, or any other single cause. Even after decades of cholesterol testing, blood-pressure screening, statins, stents, bypass surgery, and public-health campaigns, heart disease remains the country’s most persistent killer.
That makes the usual explanation worth questioning. Heart disease is often described as if it were a simple mechanical problem: cholesterol floats through the blood, sticks to the inside of the arteries, piles up over time, and eventually blocks the artery like waste inside a pipe.

That image is easy to understand, but it is not biologically adequate. An artery is not a pipe; it is living tissue. It senses the blood, responds to injury, regulates inflammation, controls clotting, contracts and relaxes, and repairs itself.
Cholesterol is present in heart disease, but cholesterol alone does not explain why arteries become inflamed, why LDL particles become oxidized, why plaques become unstable, why clots form, or why the heart muscle can lose the energy required to function.
A more complete account begins with a different premise: heart disease is not primarily a plumbing problem, but a disorder of biological regulation. It involves oxidation, inflammation, immune activity, tissue repair, clotting, mineral balance, hormonal stress, and energy production. Cholesterol participates in this process, but it is only one part of a much larger biological system.
The better question is not simply, “How much cholesterol is in the blood?” The better question is: what conditions make arteries inflamed, LDL damaged, plaques unstable, blood more prone to clotting, and the heart unable to maintain its own energy and structure?
That question reveals the central distinction: heart disease is not only a structural disease that later disrupts function. Long-running problems of function - oxidation, inflammation, weak energy production, poor repair, stress chemistry, and disturbed metabolism - gradually become structural disease.
In plain terms, the danger is not cholesterol by itself. The danger is the internal environment that damages fats, irritates arteries, weakens repair, encourages clotting, and leaves the heart muscle without the energy it needs to keep working.
The sequence is direct: the clogged-pipe image gives way to artery-wall biology; plaque becomes an immune repair structure; LDL oxidation and PUFA peroxidation move to the center; diet is judged by long-term outcomes rather than cholesterol lowering alone; and heart disease expands into energy production, stress chemistry, thyroid function, blood pressure, gut inflammation, clotting, and treatment strategy.
Arteries Are Living Organs, Not Pipes
The clogged-pipe metaphor fails because it treats the artery as passive. A pipe has no metabolism, no immune system, no repair process, and no ability to regulate its own behavior. An artery has all of those things.
An artery is made of several active layers. The innermost layer includes the endothelium, the thin cellular lining that touches the blood. The endothelium is not just a barrier; it helps control what enters the artery wall, regulates blood vessel tone, influences clotting and inflammation, and communicates with deeper layers of the vessel.
Beneath that lining is tissue that can become involved in plaque formation. The middle layer contains smooth muscle cells, which allow the artery to constrict and dilate. The outer layer provides structural support.
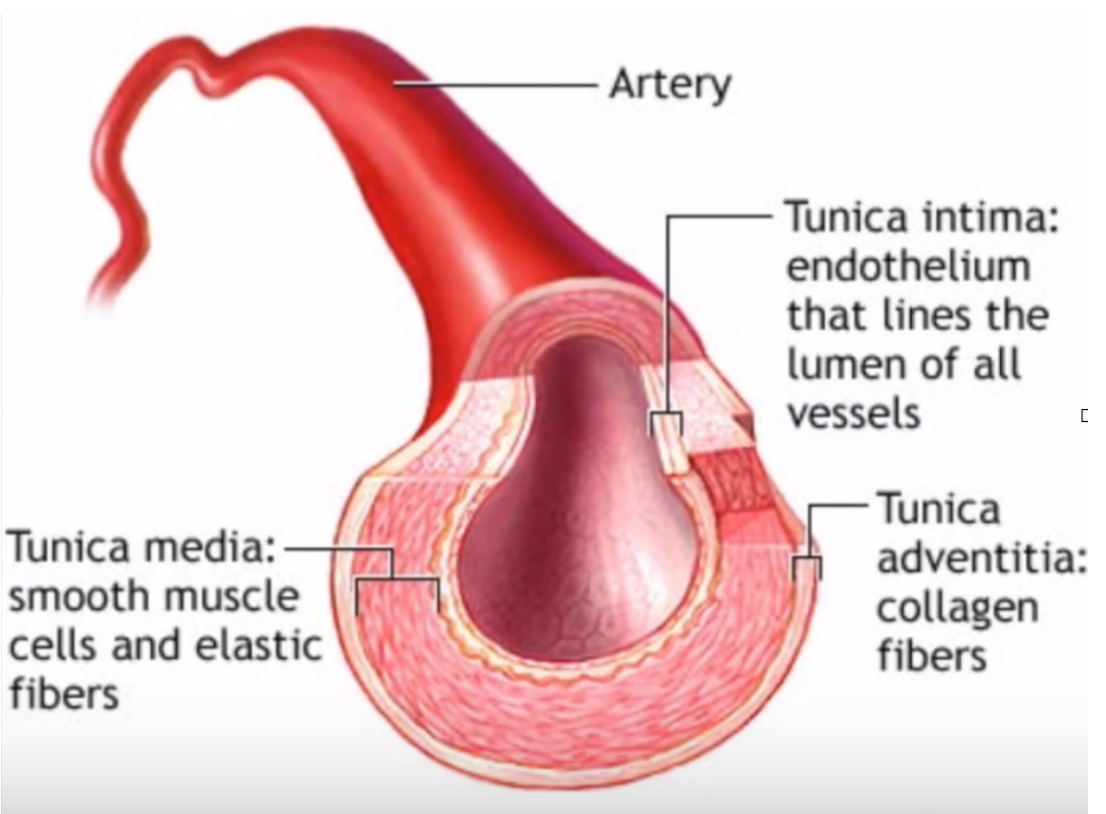
Figure 1. Artery wall anatomy, showing the endothelial lining, tunica media, and tunica adventitia.
The anatomy makes the case. The open center is the lumen, where blood moves, but the disease does not simply pile up there like sludge in a pipe. Around that opening are living layers: the tunica intima with its endothelial lining, the muscular tunica media that helps the vessel tighten and relax, and the outer adventitia, rich in connective tissue. Atherosclerosis develops in and beneath these active layers, which is why the artery has to be understood as responsive tissue, not plumbing.
This structure matters because atherosclerotic plaque does not simply form on the open inner surface of the artery like grease in a drain. Plaque develops inside the artery wall, beneath the endothelial lining.
That single fact changes the entire picture. If plaque were just debris collecting inside a tube, the disease would be mainly mechanical. But plaque forms inside living tissue, which means the disease process involves injury, immune cells, oxidized lipids, inflammation, tissue remodeling, scarring, and repair.
In a pipe, a clog forms inside the open channel. As more material accumulates, the opening becomes smaller. Early atherosclerosis does not usually work that way.
Plaque begins beneath the endothelium, inside the wall of the artery. As plaque grows, the artery may remodel outward to preserve the open space where blood flows.²² This means a person can have significant plaque development before the artery appears severely narrowed.²²,²⁹,³⁰,³¹
Plaque Grows Inside the Wall Before It Narrows the Lumen
Atherosclerotic plaque is easier to understand if its location is stated precisely. It is not simply material floating in the lumen or stacking on the inner surface of the artery. It develops beneath the endothelial lining, within the intimal space of the arterial wall, between the blood-facing endothelium and the deeper layers of the vessel.
That location changes the mechanical picture. As plaque expands, it can push outward into the surrounding vessel wall while the tissue around it pushes back. The artery then remodels as living tissue rather than behaving like a rigid tube. This is why the same amount of buildup has very different consequences in a pipe than it does in an artery.
In a pipe, material that fills 60 percent of the open channel would leave about 40 percent of the opening. Early atherosclerosis does not usually follow that pattern. As plaque burden increases, the artery can enlarge outward and preserve the lumen where blood flows. In the classic Glagov remodeling pattern, visible narrowing tends to become more obvious only after the artery’s compensatory enlargement is exceeded.²²
This means a lumen-centered image can underestimate disease burden. The opening may still look relatively preserved while plaque, oxidized lipids, immune cells, foam cells, repair tissue, and scar formation are accumulating inside the wall. The important question is not only whether the artery is narrowed, but what kind of biological process is taking place within the artery wall.
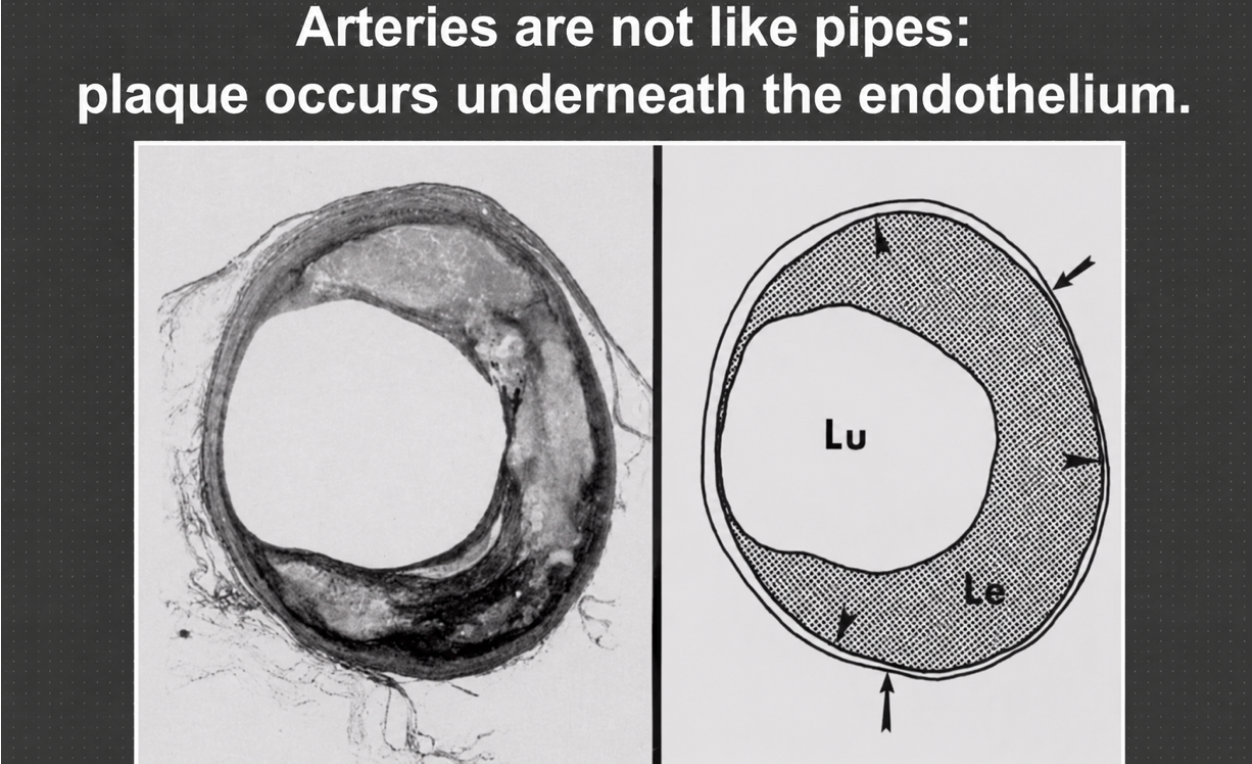
Figure 2. Atherosclerotic plaque developing beneath the endothelium rather than simply collecting inside the vessel lumen.

This image makes the pipe metaphor collapse. The plaque is not sitting neatly on the inside surface of the open channel; it is embedded in the artery wall beneath the endothelial lining. The left side shows a real cross-section, while the right side turns the same idea into a simplified diagram, with the lumen still open in the center. That is the crucial point: an artery can remodel outward and preserve blood flow for a time even while a serious biological process is developing in the wall.
This outward remodeling is one of the clearest reasons the pipe metaphor fails. The artery can enlarge to accommodate the growing plaque, so the lumen may remain relatively open during much of the early disease process. Literal narrowing of the blood channel tends to become more obvious later, after plaque accumulation, repeated injury, inflammation, scarring, and structural remodeling have advanced far enough.
The problem is not simply that cholesterol fills the artery from the inside. The problem is that the artery wall becomes a site of biological damage and failed repair.
LDL enters the artery wall; the polyunsaturated, or PUFA-rich, parts of LDL become damaged in an oxidizing environment; the immune system responds; macrophages enter; foam cells form; inflammatory material accumulates; and the body tries to contain the damage with a fibrous cap. If the cap weakens and ruptures, a clot can form.¹⁸,¹⁹,²⁰,²¹,²⁵,²⁶
That is not passive buildup. It is an active disease process occurring inside living tissue.
Plaque Is an Immune Repair Structure
Atherosclerotic plaque is often described as fat buildup, but that description is too simple. Plaque contains cholesterol, but it also contains immune cells, oxidized lipids, dead cells, calcium, collagen, and inflammatory material.²¹ In many ways, plaque can be understood as an immune containment structure: the body forms it in an attempt to contain damaged and dangerous material inside the artery wall.
The key damaged material is often oxidized LDL. More specifically, the most oxidation-prone material inside LDL is PUFA. Cholesterol may be present in the particle, but PUFA is the part most vulnerable to lipid peroxidation.¹⁸ That makes PUFA central to the transformation of ordinary LDL into damaged, inflammatory LDL.
LDL particles can enter the space beneath the endothelium.²⁵ If they remain there long enough, and if the local environment is oxidizing, they can become chemically damaged. Once LDL is oxidized, the body no longer treats it like normal LDL. Oxidized LDL can injure endothelial cells and attract immune cells.¹⁸,¹⁹,²⁰
Macrophages enter the artery wall to remove oxidized LDL. These macrophages take up oxidized LDL through scavenger receptors. Unlike normal LDL receptor activity, this process is poorly controlled. A macrophage can continue consuming oxidized LDL until it becomes overloaded. At that point, it becomes a foam cell.¹⁹,²¹
Foam cells are central to plaque formation. Over time, they may die and release their contents. This creates a necrotic core, which can contain oxidized lipids, dead cell material, inflammatory substances, calcium, and debris.
The body then tries to cover this dangerous core with a fibrous cap made largely of collagen-rich tissue, with smooth muscle cells helping build and maintain it. Its purpose is protective: it separates the inflammatory contents of the plaque from the bloodstream.²⁶
A plaque with a strong cap may remain stable. A plaque with a weak cap may rupture. This is why the danger is not only how much plaque exists. The danger is whether the plaque is stable or unstable.²⁶
A heart attack is not always caused by an artery slowly narrowing until blood can no longer pass. Many heart attacks occur when a plaque ruptures. When the fibrous cap breaks, the inflammatory contents of the plaque are exposed to the blood. The blood reacts as if there has been an injury: platelets activate, and a clot forms.
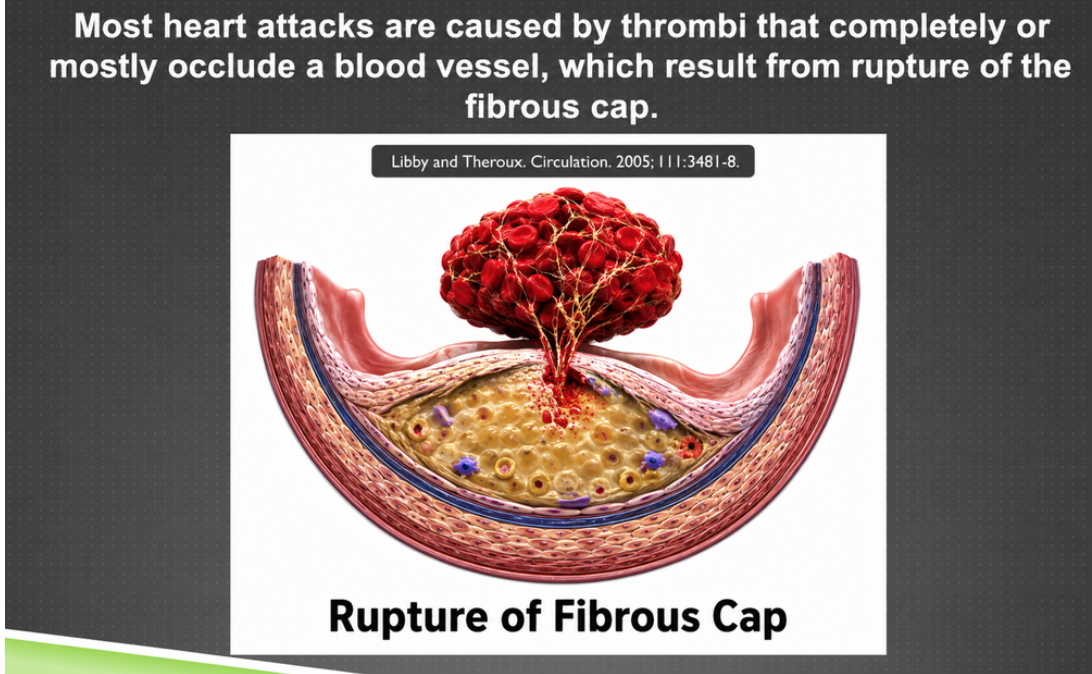
Figure 3. Fibrous-cap rupture, in which the plaque core is exposed to blood and can trigger clot formation.
Here the story moves from chronic containment to acute danger. The pale material under the cap represents the inflammatory plaque core, and the fibrous cap is the cover that keeps that material separated from the bloodstream. When the cap ruptures, blood encounters the plaque contents as if it has found a wound. Platelets and clotting factors respond, and a thrombus can form quickly. A heart attack is often not the slow final closing of a pipe but a sudden rupture-and-clot event.
If that clot blocks a coronary artery, part of the heart muscle can be deprived of oxygen, causing a heart attack. If a similar process happens in an artery supplying the brain, it can contribute to an ischemic stroke.
The immediate danger is often not just plaque size. It is plaque instability plus clot formation. A smaller unstable plaque can be more dangerous than a larger stable plaque, while a plaque that remains contained may cause less acute danger than one with a thin, inflamed, weakened cap that suddenly ruptures.
That is why heart disease cannot be understood only by asking whether an artery is narrowed. The deeper questions are whether the artery wall is inflamed, whether the plaque is structurally stable, and whether the blood is prone to clotting.
Narrowing Can Reflect Repeated Injury and Repair
Severe narrowing can develop through repeated cycles of rupture and healing.²³,²⁴ A plaque may rupture in a small, subclinical way. The rupture may heal. Scar tissue may form. Then a new plaque layer may develop on top of the old one. Over time, this repeated injury-and-repair process can narrow the artery.²³,²⁴
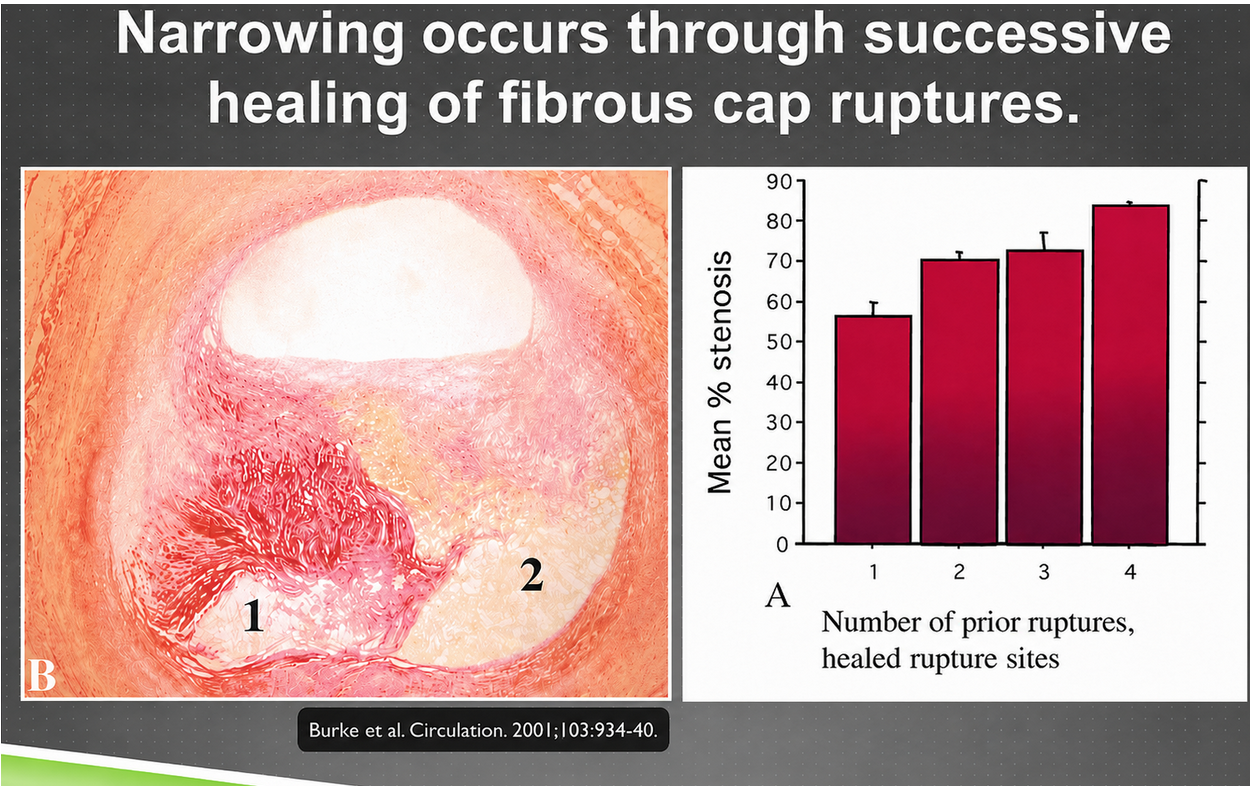
Figure 4. Progressive arterial narrowing through repeated cycles of fibrous-cap rupture, healing, and scarring.
The left side shows the layered history that a mature plaque can carry: injury, healing, scar tissue, and new plaque formation stacked over time. The bar chart translates that history into stenosis, meaning narrowing of the artery opening. As the number of prior healed ruptures rises, the average narrowing rises too. The image is important because it changes the mental picture from one smooth cholesterol mass growing inward to repeated biological damage followed by imperfect repair.
This is different from the idea of one mass slowly growing inward like sludge in a pipe. The early plaque may push outward into the artery wall, while later narrowing can reflect layer after layer of healed rupture, scar tissue, and new plaque formation.
Stenosis is often a history of repeated, incomplete resolution. The body identifies injury, attempts containment, repairs the area, scars, and repeats the process. Eventually, the repair process itself can contribute to narrowing and instability.
That distinction matters. It shifts the focus from “what is sticking to the artery?” to “why is the artery repeatedly injured, inflamed, rupturing, healing, and unable to resolve the damage cleanly?”
LDL Is Not Simply “Bad Cholesterol”
LDL is often called “bad cholesterol,” but that phrase hides what LDL actually is. LDL is not cholesterol itself. LDL is a lipoprotein particle. It transports cholesterol, triglycerides, fat-soluble vitamins, coenzyme Q10, and other fat-soluble substances through the blood.
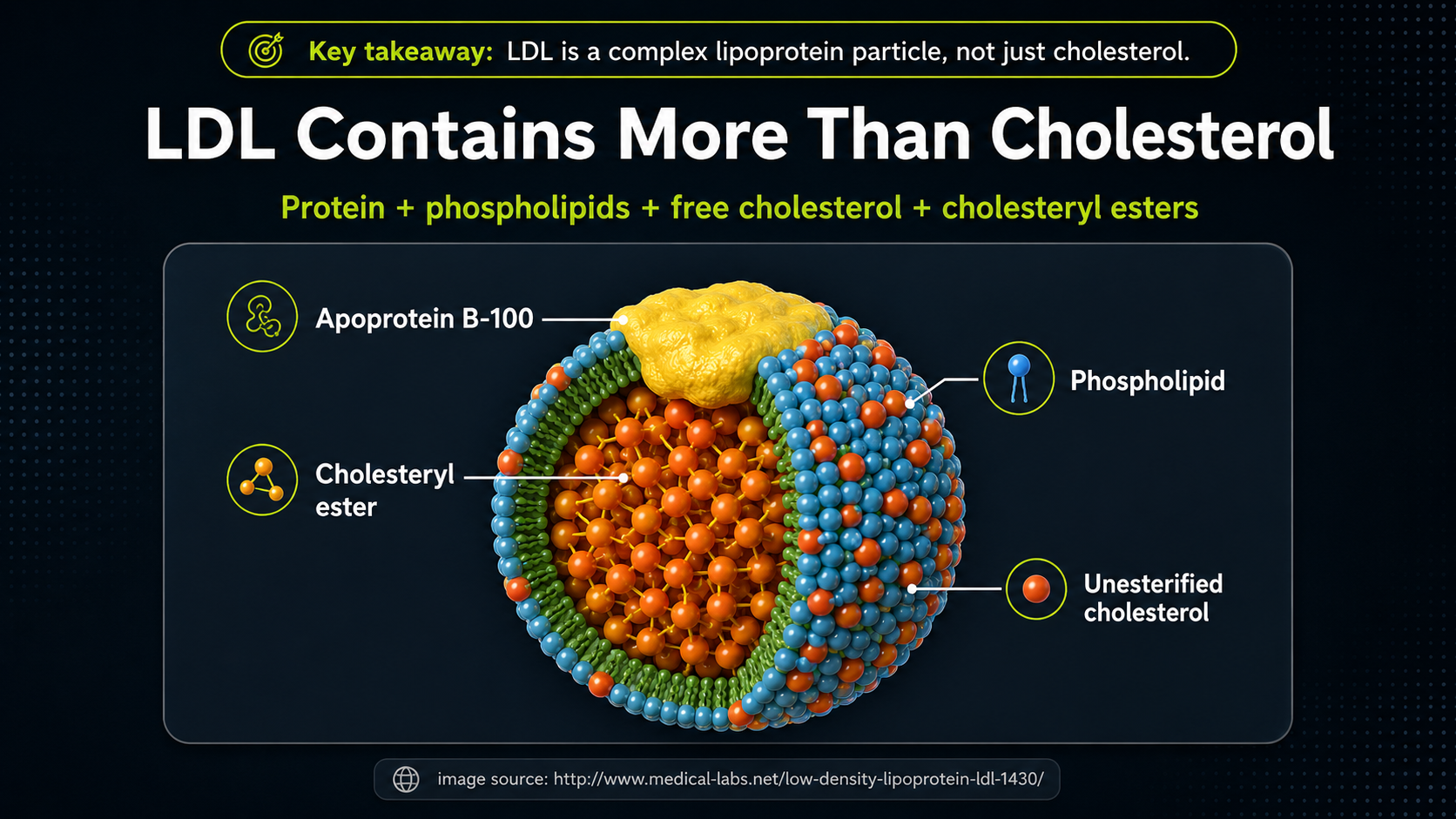
Figure 5. LDL particle structure, showing that LDL carries more than cholesterol.
This cutaway view shows why LDL should not be imagined as a blob of cholesterol. It is a structured transport particle. Apoprotein B-100 is the large identifying protein on the surface; the outer shell contains phospholipids and unesterified cholesterol; the inner core carries cholesteryl esters and other fat-soluble cargo. The whole particle matters, not only the cholesterol number. The phospholipid surface is especially important because its fatty-acid composition can include PUFA, making the particle more vulnerable to oxidation.
That distinction matters because the biological behavior of LDL depends on the whole particle: its protein structure, lipid membrane, antioxidant protection, fatty acid composition, circulation time, and tendency to become oxidized.
Normal LDL can be cleared through regulated LDL receptors. Oxidized LDL is handled differently. It is taken up by macrophages through scavenger receptors, which can lead to foam-cell formation.
LDL concentration alone does not explain the disease. LDL becomes dangerous in the context of entry, retention, oxidation, endothelial injury, and immune activation. The central issue is not simply LDL. The central issue is oxidized LDL in an inflamed artery wall.
The surface membrane of LDL is made partly of phospholipids, and many of the fatty acids in those phospholipids can be polyunsaturated fatty acids, or PUFAs. That detail is not incidental.
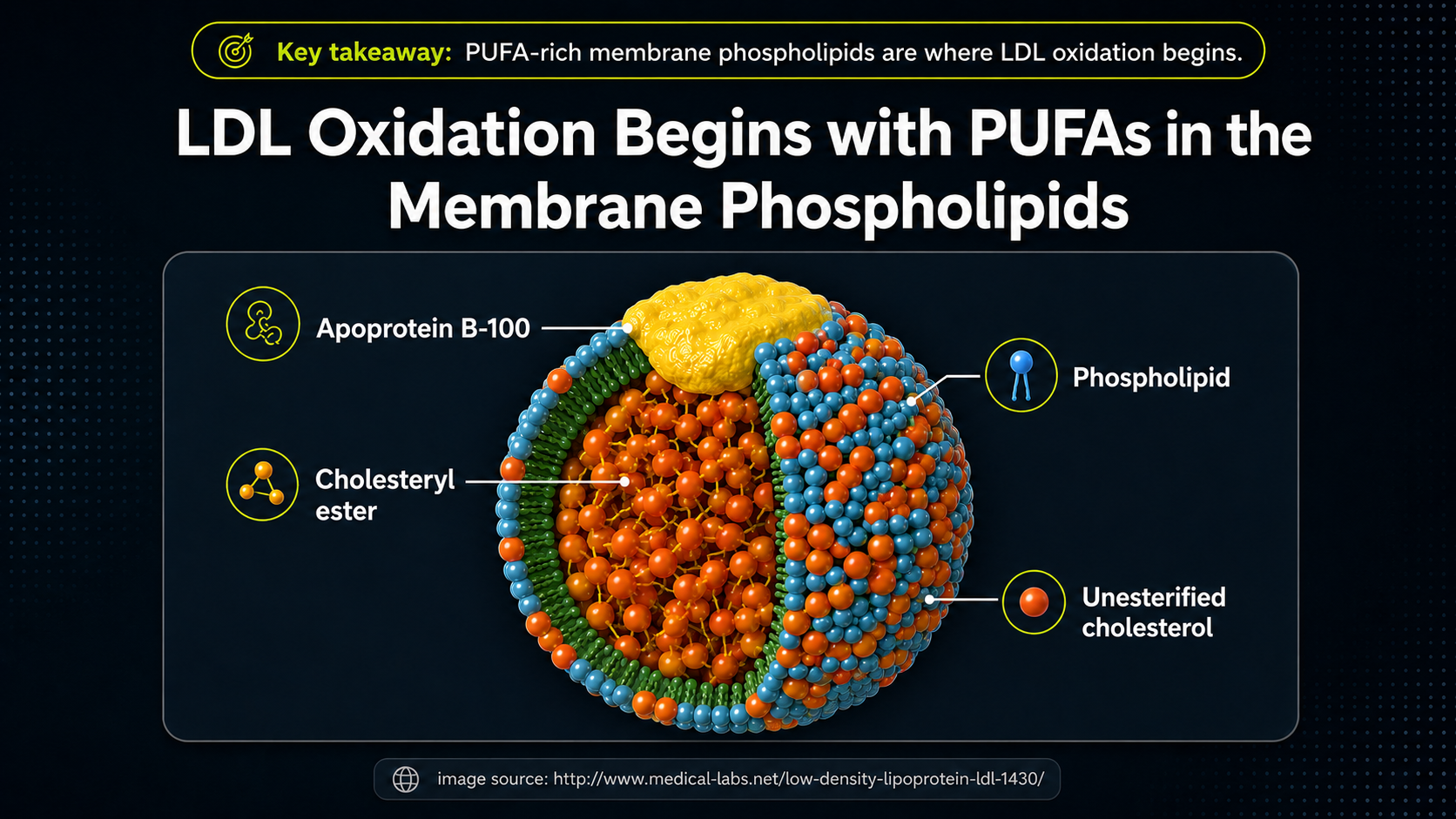
Figure 6. PUFA-containing phospholipids in the LDL membrane, highlighting why fatty-acid composition matters for oxidation.
The same LDL particle now becomes a more specific chemical problem. The phospholipid layer is the surface that meets the blood and the artery-wall environment, and the fatty-acid tails in that layer can include PUFA. That detail connects LDL biology to lipid peroxidation. Oxidation does not have to begin with cholesterol itself; it can begin in the PUFA-containing membrane lipids and then change how the entire LDL particle behaves.
PUFA is the unstable material that turns oxidative stress into lipid peroxidation. Saturated fats are comparatively resistant to this kind of oxidative damage. PUFAs, because of their chemical structure, are much easier to damage through oxidation.
When the PUFA-rich parts of LDL become oxidized, the LDL particle changes. It becomes more inflammatory and more likely to be taken up by immune cells.
Oxidized LDL can injure the endothelium, attract macrophages, promote foam-cell formation, and contribute to plaque development. This is why PUFA is not merely another dietary fat. It is the material that makes LDL more vulnerable to becoming a signal of injury.
The bar chart gives the mechanism some bite. Along the bottom is the concentration of oxidized LDL, and along the side is endothelial-cell viability. At low concentrations, the cells remain mostly viable; at higher concentrations, especially toward the far right, viability drops sharply. In other words, oxidized LDL is not just a marker that happens to appear near disease. In this experimental setting, enough oxidized LDL can directly injure the cells that line the blood vessel.

Figure 7. Oxidized LDL and endothelial injury, linking LDL modification to vascular inflammation.
This helps explain why LDL modification matters more than LDL as an isolated number. Native LDL is normally handled through regulated LDL receptors, but when LDL is oxidized, its behavior changes.
It can attract immune cells, lose normal recognition through the LDL receptor system, and become a target for scavenger receptors on macrophages. LDL receptor uptake is regulated; scavenger receptor uptake is not controlled in the same way. A macrophage taking up oxidized LDL can continue until it becomes overloaded and turns into a foam cell.
The process begins when native LDL enters the subendothelial space.²⁵ If the environment is oxidizing, PUFAs in the LDL membrane begin to peroxidize.¹⁸ Early oxidation products signal endothelial cells to attract monocytes, which enter the artery wall and become resident macrophages.¹⁹,²⁰
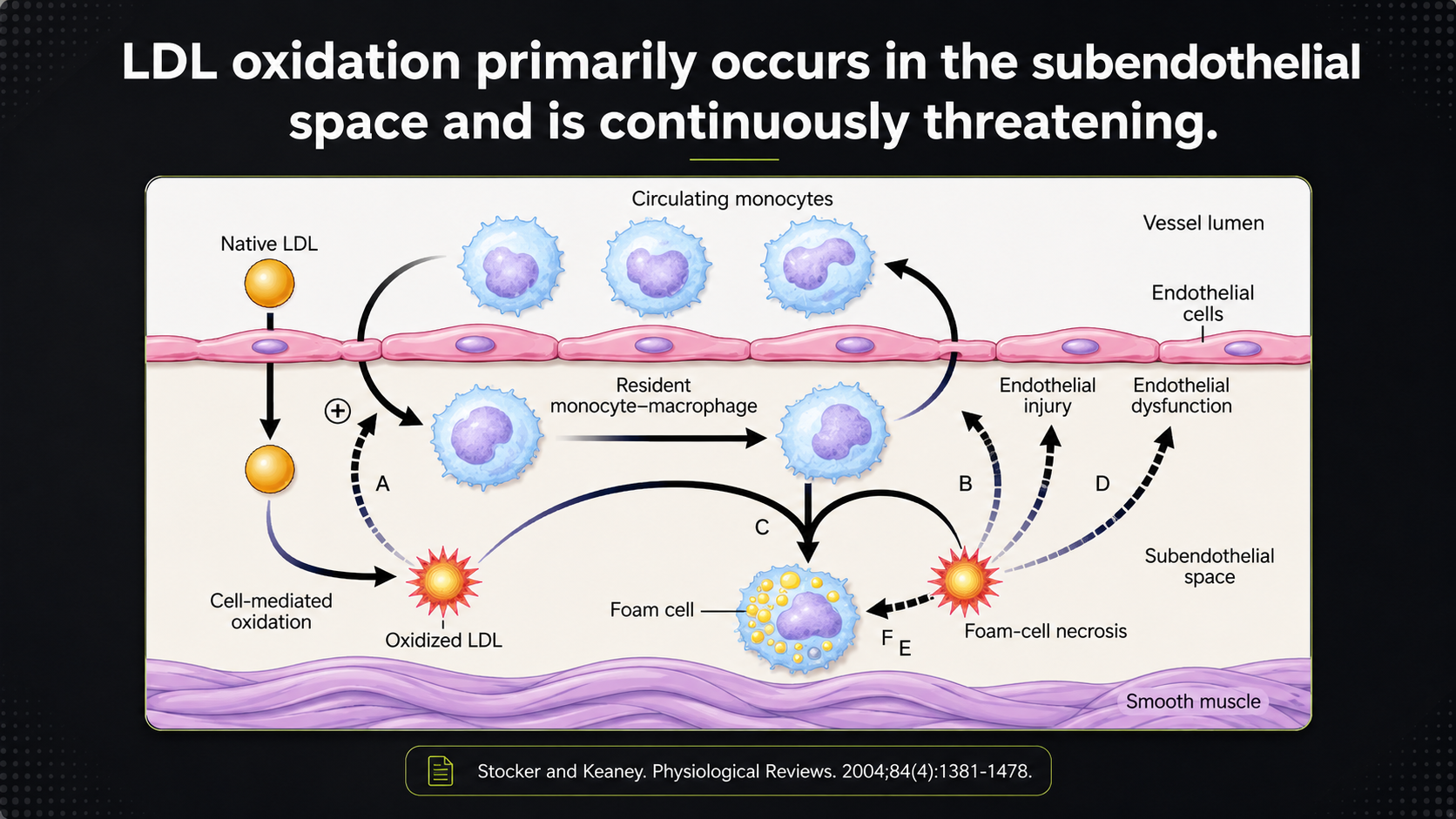
Figure 8. LDL oxidation in the subendothelial space and the macrophage/foam-cell response that follows.
This diagram follows LDL after it slips beneath the endothelium into the subendothelial space. Native LDL can oxidize there, especially when the local environment is inflammatory or oxidizing. Oxidized LDL then attracts monocytes, which enter the artery wall and become macrophages. Those macrophages take up oxidized LDL and can turn into foam cells. The arrows are not decoration; they show a self-reinforcing cycle in which oxidation injures the lining, injury makes the wall more permeable and inflamed, and that invites still more LDL and immune activity into the same place.
As LDL oxidation advances, the particle is recognized and cleared differently. Macrophages take up oxidized LDL through scavenger receptors, become foam cells, and eventually die. Their death releases oxidized lipid material, enlarges the necrotic core, increases inflammation, and makes the fibrous cap more vulnerable.¹⁸,¹⁹,²¹
This creates a self-reinforcing cycle: LDL enters the artery wall, becomes oxidized, injures the endothelium, makes the endothelium more permeable and inflamed, invites more LDL and immune cells into the area, drives macrophages into foam-cell formation, and enlarges the plaque core as foam cells die.¹⁸,¹⁹,²⁰,²¹,²⁵
This process explains heart disease more completely than the idea that cholesterol simply sticks to the artery wall. It also explains why antioxidant defense matters.
The body must prevent PUFA peroxidation in circulating LDL and inside the artery wall. If antioxidant defenses are weak and inflammation is high, LDL is more likely to become damaged.
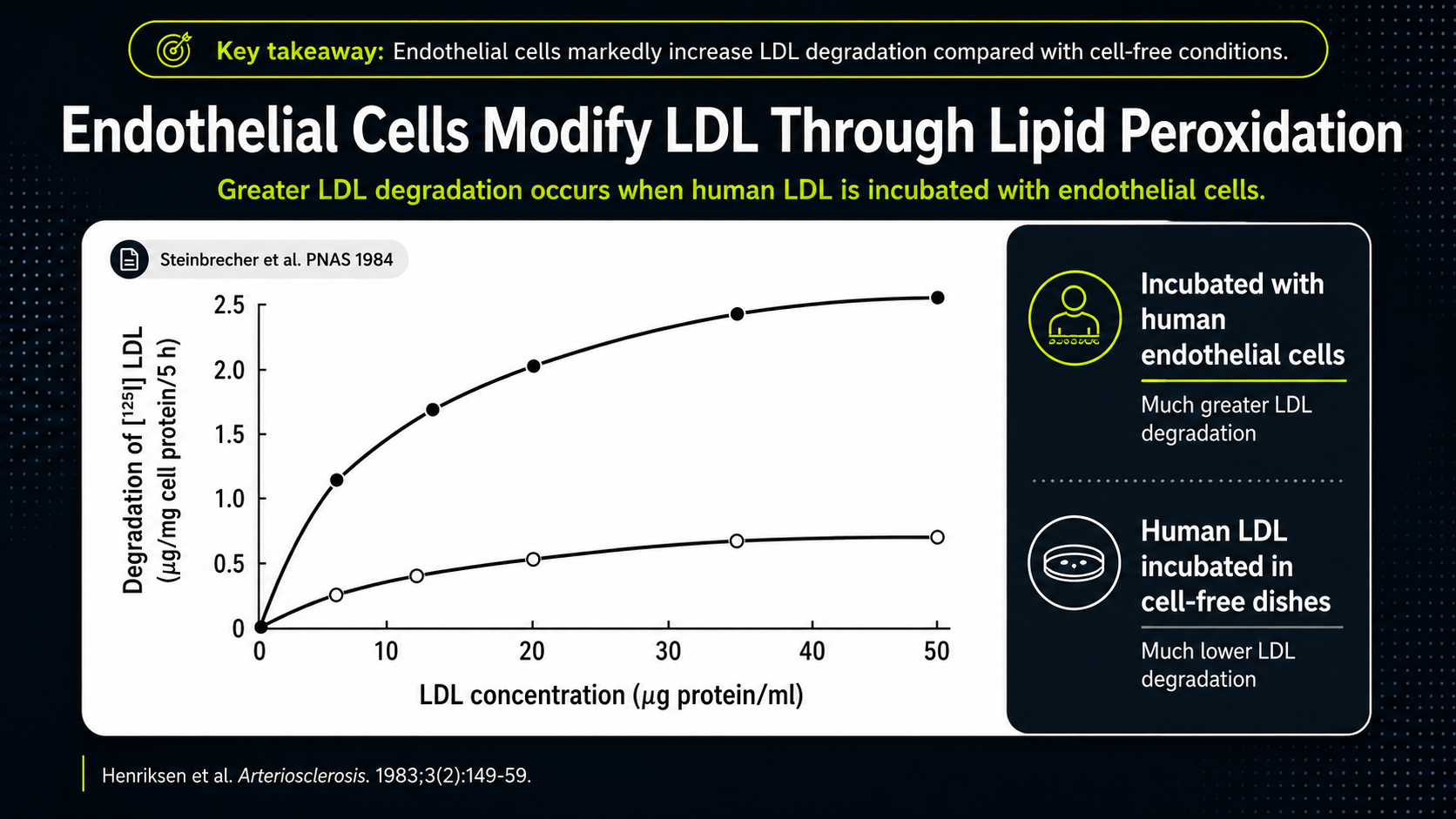
Figure 9. LDL phospholipid oxidation over time, illustrating the need for antioxidant protection.
The graph shows LDL phospholipids being degraded when LDL is exposed to endothelial cells. The rising line is the key: human LDL incubated with endothelial cells undergoes much more phospholipid degradation than LDL kept in cell-free dishes. The artery-wall environment can actively modify LDL. That makes LDL oxidation a local biological process involving cells, oxidative chemistry, and vulnerable membrane lipids, not merely cholesterol drifting unchanged through the bloodstream.
Antioxidant Defense and Tissue Repair Decide Whether Plaque Stays Stable
This is where nutrition enters the story most directly. If oxidized LDL drives plaque formation, and PUFA is the most oxidation-prone material inside that LDL, then antioxidant defense is central to cardiovascular health.
The issue is not only how much cholesterol is present. The issue is how much oxidation-prone PUFA is present, whether those lipids are protected from oxidation, and whether the artery wall can repair itself properly.
Vitamin E helps protect lipids from oxidation. Vitamin C supports antioxidant recycling and collagen synthesis. Glutathione helps control oxidative stress.
B vitamins, minerals, ATP production, and NADPH production all support the broader antioxidant defense system. NADPH is especially important because cells use it to keep antioxidant systems recycled and active.
LDL particles require antioxidant protection while they circulate. The liver first produces lipoproteins in a form that eventually gives rise to LDL, and the antioxidant resources available at that time help determine how well those particles are protected during their life in the blood.
Once antioxidant protection is depleted, the particle becomes more vulnerable to oxidation.
The artery wall also requires protection. If the subendothelial space is inflamed and oxidizing, LDL is more likely to become damaged there.
Inflammation creates oxidative stress, and oxidative stress creates more inflammation. Atherosclerosis develops inside that loop.
LDL clearance and LDL retention also matter. Thyroid hormone supports LDL receptor activity, helping remove LDL particles from the blood before they have more time to oxidize. Inflammation tends to push in the opposite direction by impairing normal regulation and making the artery wall more permeable.
Retention inside the artery wall is another key step. LDL becomes more dangerous when it gets stuck in the subendothelial space long enough to oxidize.
The stickiness of that space is influenced by proteoglycans, which are protein-sugar structures in the artery wall.²⁵ Nutrients involved in proteoglycan regulation, including manganese, may therefore matter because a stickier subendothelial space can give LDL more time to oxidize.
Plaque stability also depends on tissue repair. The fibrous cap protects the bloodstream from the inflammatory core of the plaque. That cap depends heavily on collagen.²⁶
Inflammation can degrade collagen. The body must continually synthesize enough collagen to maintain the cap.²⁶ Vitamin C, copper, glycine, adequate protein, and healthy energy production all matter for collagen formation and tissue maintenance.
If collagen breakdown exceeds collagen repair, the cap weakens. A weak cap is more likely to rupture.
Calcification also matters. Calcium can make plaque tissue more rigid or brittle. Vitamins A, D, and K-dependent proteins are involved in calcium regulation and tissue remodeling.²⁷,²⁸
Plaque stability depends on more than cholesterol. It depends on inflammation, collagen synthesis, mineral regulation, fat-soluble vitamins, antioxidant status, LDL clearance, LDL retention, and the body’s ability to repair tissue.
A plaque becomes dangerous when its structure can no longer contain its inflammatory contents. PUFA contributes to that danger in two related ways: it can oxidize inside LDL and it can serve as the raw material for inflammatory lipid mediators that keep the vascular injury active.
Cholesterol May Participate in Containment, Not the Original Injury
Cholesterol is present in plaque, but its presence does not prove that cholesterol started the disease.
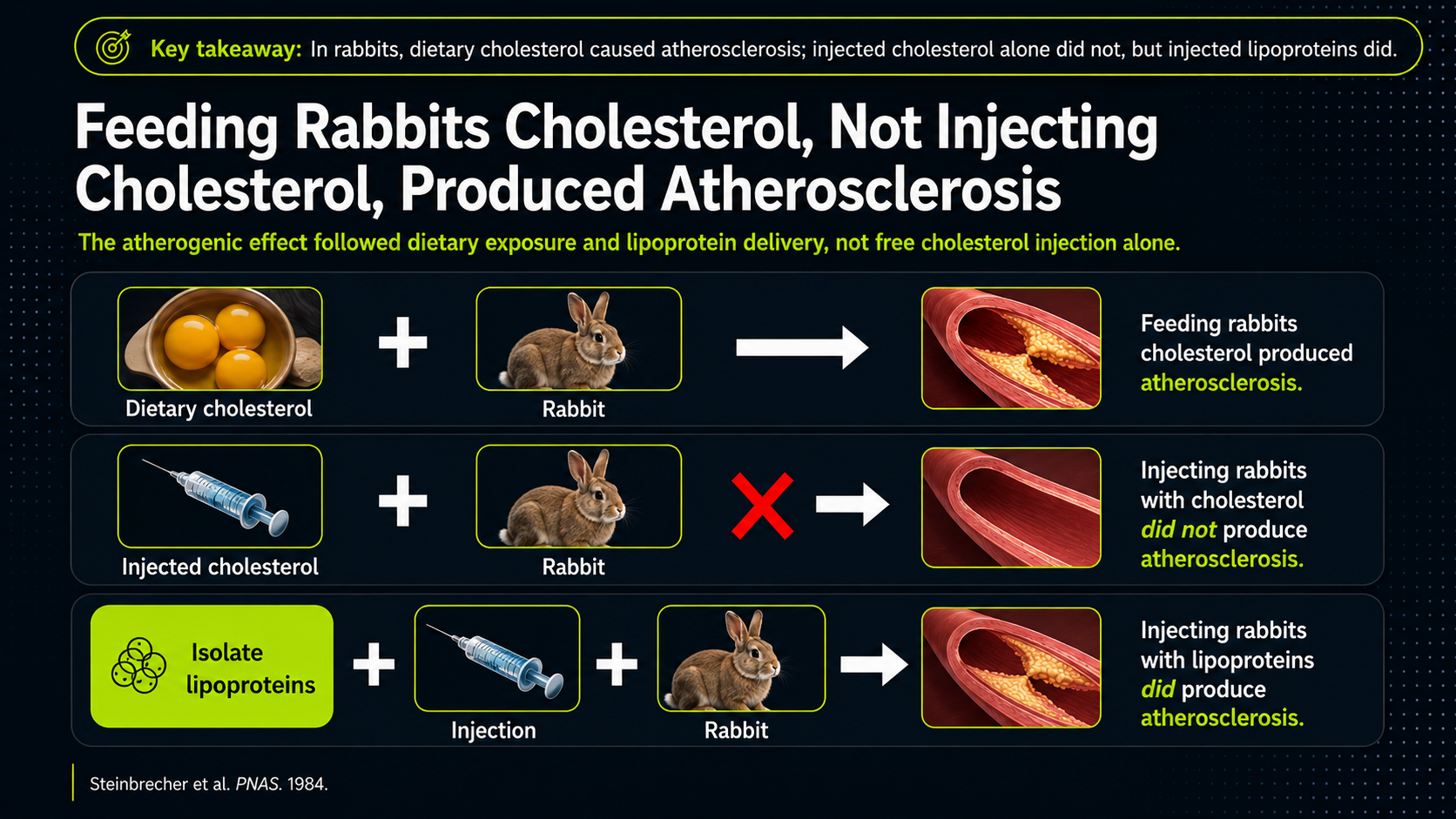
Figure 10. Cholesterol in context: experimental examples suggesting that cholesterol’s effect depends on the biological environment surrounding lipoproteins.
The three rows are meant to separate cholesterol from the biological context that carries it. Feeding cholesterol through digestion produced atherosclerosis in this rabbit model; simply injecting cholesterol did not, as the red X emphasizes; injecting lipoproteins did. The takeaway is not a literal dietary rule from rabbits. It is a conceptual point: cholesterol's effect changes depending on its carrier and environment. The danger belongs less to cholesterol floating by itself than to lipoprotein biology inside living tissue.
Plaque also contains immune cells, oxidized lipids, dead cells, calcium, collagen, and inflammatory material. Cholesterol appears in an environment already shaped by injury, oxidation, immune activity, and repair.
A more complete interpretation is that cholesterol can be part of the body’s attempt to repair or contain damage. The body brings materials to an injured site in an effort to stabilize it. Cholesterol may participate in that response.
This does not mean cholesterol is irrelevant. It means cholesterol should not be isolated from the broader disease process or treated as the original injury by default.
Cholesterol also sits inside several protective systems. The body uses it to make bile acids and steroid hormones, and lipoproteins themselves can participate in innate defense by binding irritating bacterial products in the blood.
Seen from that angle, cholesterol is not just a number to lower or a substance found in plaque. It is part of a larger repair, transport, hormone, and defense network. The question is whether that network is working cleanly or being forced into chronic damage control.
The better question is not simply, “How should cholesterol be removed from the blood?” The better question is, “Why did the artery wall become inflamed, oxidizing, and unable to repair itself normally?”
Diet Cannot Be Judged by Cholesterol Lowering Alone
The dietary question is where the cholesterol-centered explanation runs into one of its clearest problems. If heart disease were mainly a cholesterol-storage problem, then a diet that lowers cholesterol should be assumed to help.
But if the disease process depends on PUFA oxidation, inflammatory lipid mediators, plaque instability, clotting, and failed repair, then cholesterol lowering is only one piece of the story.¹⁵,¹⁶
The standard dietary case is often presented simply: saturated fat raises cholesterol, polyunsaturated fat lowers cholesterol, and therefore saturated fat is harmful while polyunsaturated fat is protective. That case depends heavily on cholesterol lowering as the main measure of success.
But if oxidized LDL is central to atherosclerosis, then cholesterol lowering alone is not enough. A diet could lower cholesterol while increasing the oxidation-prone PUFA inside lipoprotein membranes. In that case, the cholesterol number might improve while the underlying oxidative problem remains unresolved or becomes worse.
Polyunsaturated fatty acids are more vulnerable to lipid peroxidation than saturated fats. They are also precursors to inflammatory mediators, including those derived from arachidonic acid, which is produced from linoleic acid, the major omega-6 PUFA in many vegetable oils. That means PUFA-rich diets cannot be judged only by their effect on cholesterol.
This is also why the problem cannot be treated as a short-term oil swap. Unsaturated fats can be stored in the body and released later during stress, illness, fasting, or heavy exertion.
Lowering PUFA exposure is therefore not only about what happens after one meal. It is a long-term change in the body’s available inflammatory and oxidation-prone substrate.
The real question is broader: does the dietary change reduce lipid peroxidation, inflammatory mediator production, heart attacks, strokes, plaque instability, oxidative stress, cancer risk, total mortality, and long-term disease burden? That is why the old diet trials matter. They are not historical trivia; they test whether a cholesterol-lowering dietary strategy actually improves the outcomes people care about.
One type of evidence comes from observational studies, where researchers compare people who eat more PUFA with people who eat less PUFA and then track heart-disease outcomes. Some of these studies associate higher PUFA intake with lower heart disease risk, but this kind of evidence is vulnerable to healthy-user bias.¹⁷
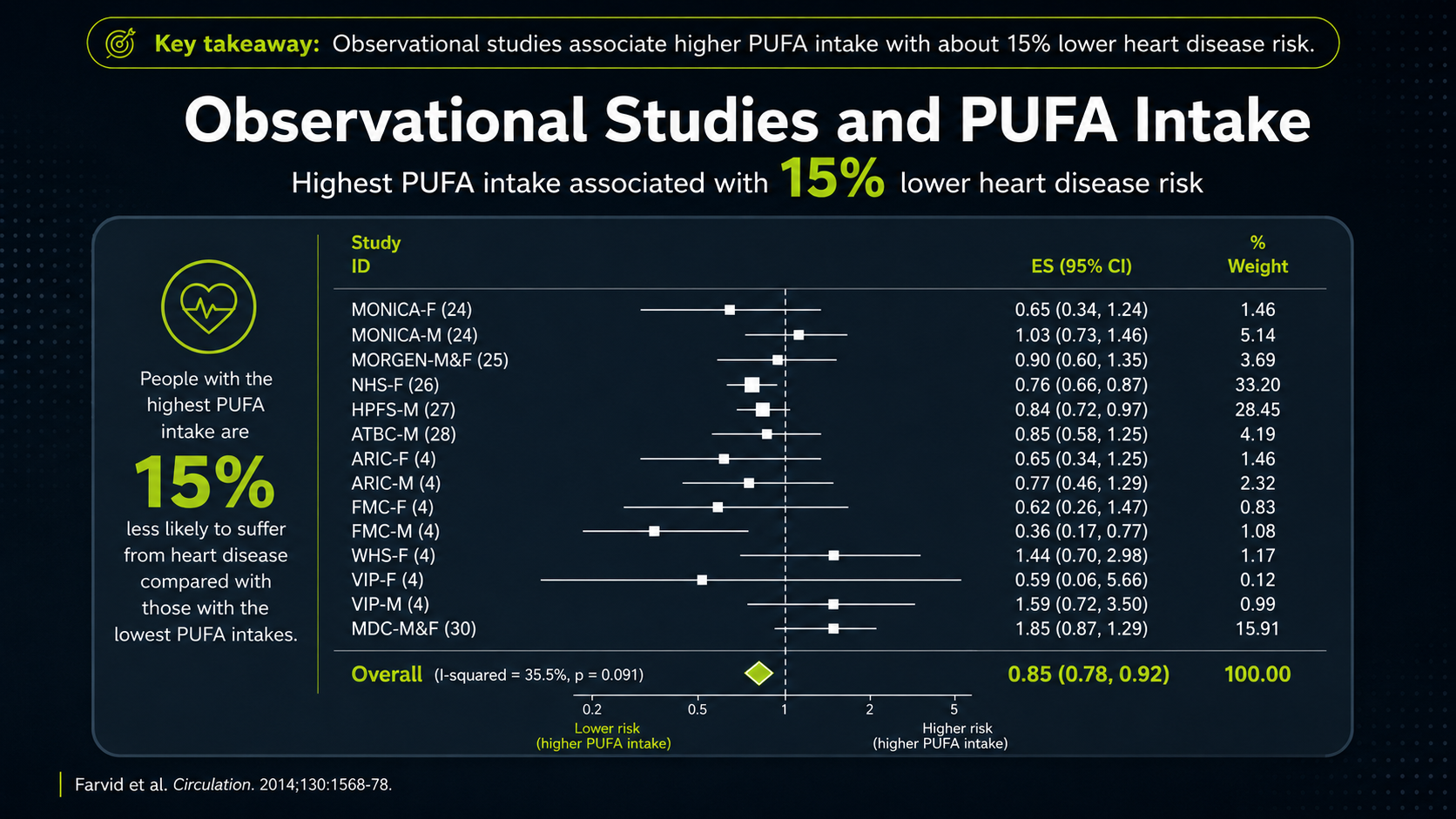
Figure 11. Observational PUFA evidence and the healthy-user-bias problem.
The forest plot compresses many observational studies into one picture. Each horizontal line is a study's confidence interval, and the square or point on that line is the study's best estimate. The larger the shaded box, the more weight that study carries in the pooled result. The vertical line at 1 means no difference; points to the left suggest lower risk with higher PUFA intake, while points to the right would suggest higher risk. The diamond at the bottom is the pooled estimate, roughly a 15 percent lower CHD-risk association. That sounds favorable, but it is still an association, not proof of cause.
That distinction matters because the people eating more PUFA in observational studies may also be the people most likely to follow health advice generally. For decades, public-health messaging told health-conscious people to replace saturated fat with vegetable oils. Those same people may also have smoked less, exercised more, eaten more fruits and vegetables, and received more medical care. A modest association can therefore reflect an entire health-conscious pattern rather than the independent effect of PUFA.
So the figure is useful, but it is not decisive. It shows why PUFA looked plausible from population data, and it also shows why that evidence cannot settle the question. A more direct test has to move from observational patterns to controlled trials and ask exactly what those trials changed.
Decades of public messaging encouraged health-conscious people to replace saturated fat with PUFA-rich oils. People following that advice may also have been more likely to exercise, smoke less, eat more fruits and vegetables, get more medical care, and follow health advice generally. That makes it difficult to isolate the effect of PUFA itself.
The reported observational advantage is also not large enough to settle the question. A modest association between higher PUFA intake and lower heart disease risk cannot prove that PUFA caused the benefit, especially when the behavior was already tied to a broader health-conscious identity.
Randomized trials can be more useful because researchers assign people, or sometimes institutions, to different diets. But even these trials have to be read carefully. The question is not only whether a trial appears favorable or unfavorable. The question is what the trial actually tested.¹⁵,¹⁶
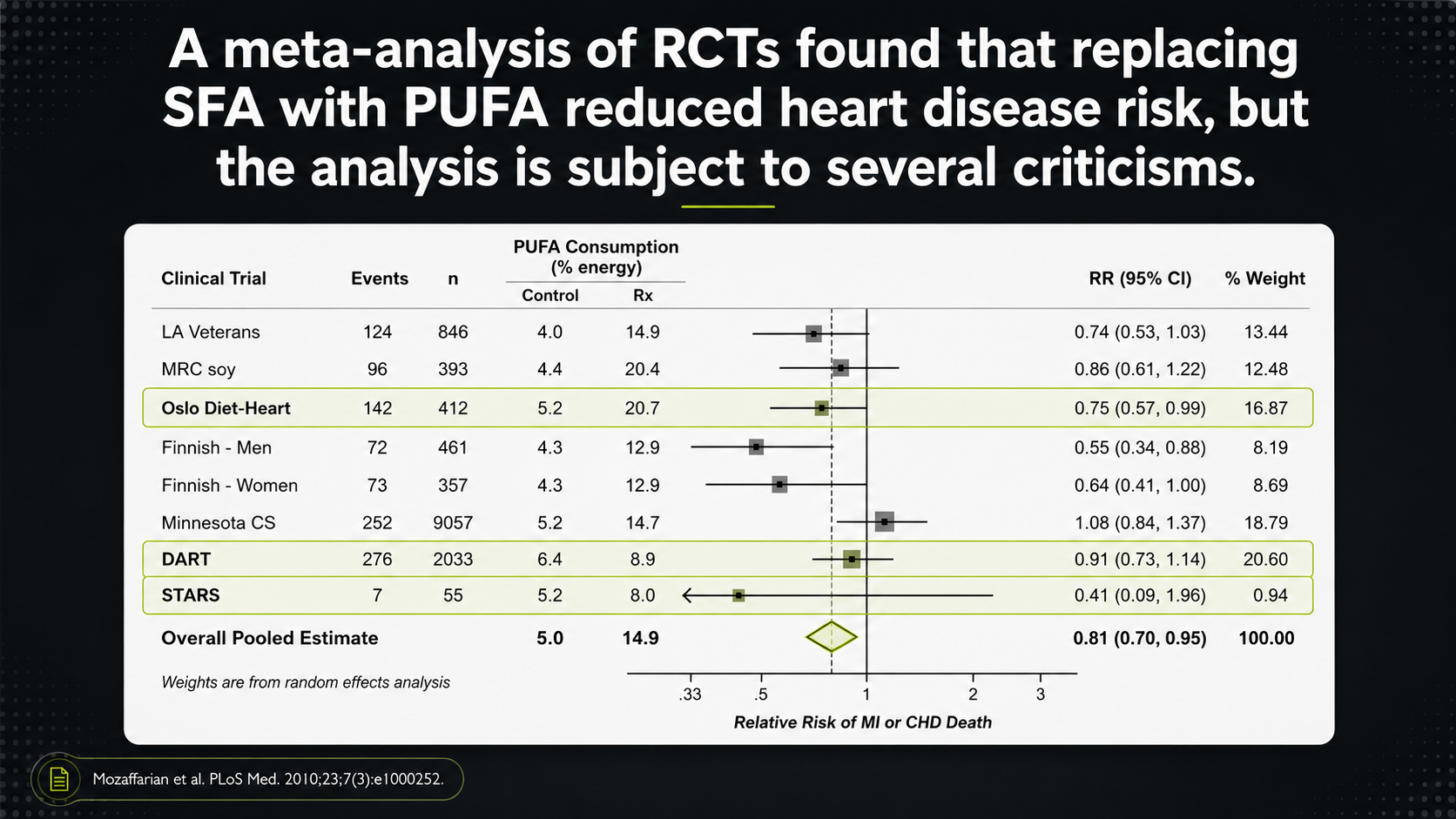
Figure 12. RCT meta-analysis framing and the importance of trial details.
The randomized-trial forest plot uses the same visual grammar, but now the stakes are higher because trials are supposed to show more about cause and effect. The right-hand weight column shows which studies pull the pooled result most strongly. A trial with a large weight can shape the conclusion far more than several smaller trials, so the first question is not simply what the diamond says. It is which trials created that diamond.
That is why trial details matter. A pooled result can look clean while the studies underneath it are messy: some may be multifactorial diet programs rather than simple fat swaps; some may have design problems; and some may be excluded even though their results would complicate the conclusion. The forest plot is therefore a map of the debate, not the end of it.
One frequently cited study is known as the Finnish Mental Hospitals Study. It compared diets in two mental hospitals over two time periods. For several years, one hospital used a diet higher in polyunsaturated fat while the other used a diet higher in saturated fat; then the hospitals switched diets.
The problem is that the individual patients were not randomly assigned in the way people may imagine when they hear “randomized trial.” The hospital was effectively the unit of assignment, and hospital-level factors such as medications, staffing, patient differences, and time-period effects could influence the results. That makes the study difficult to treat as clean evidence that PUFA itself caused the reported benefit.⁶,⁷
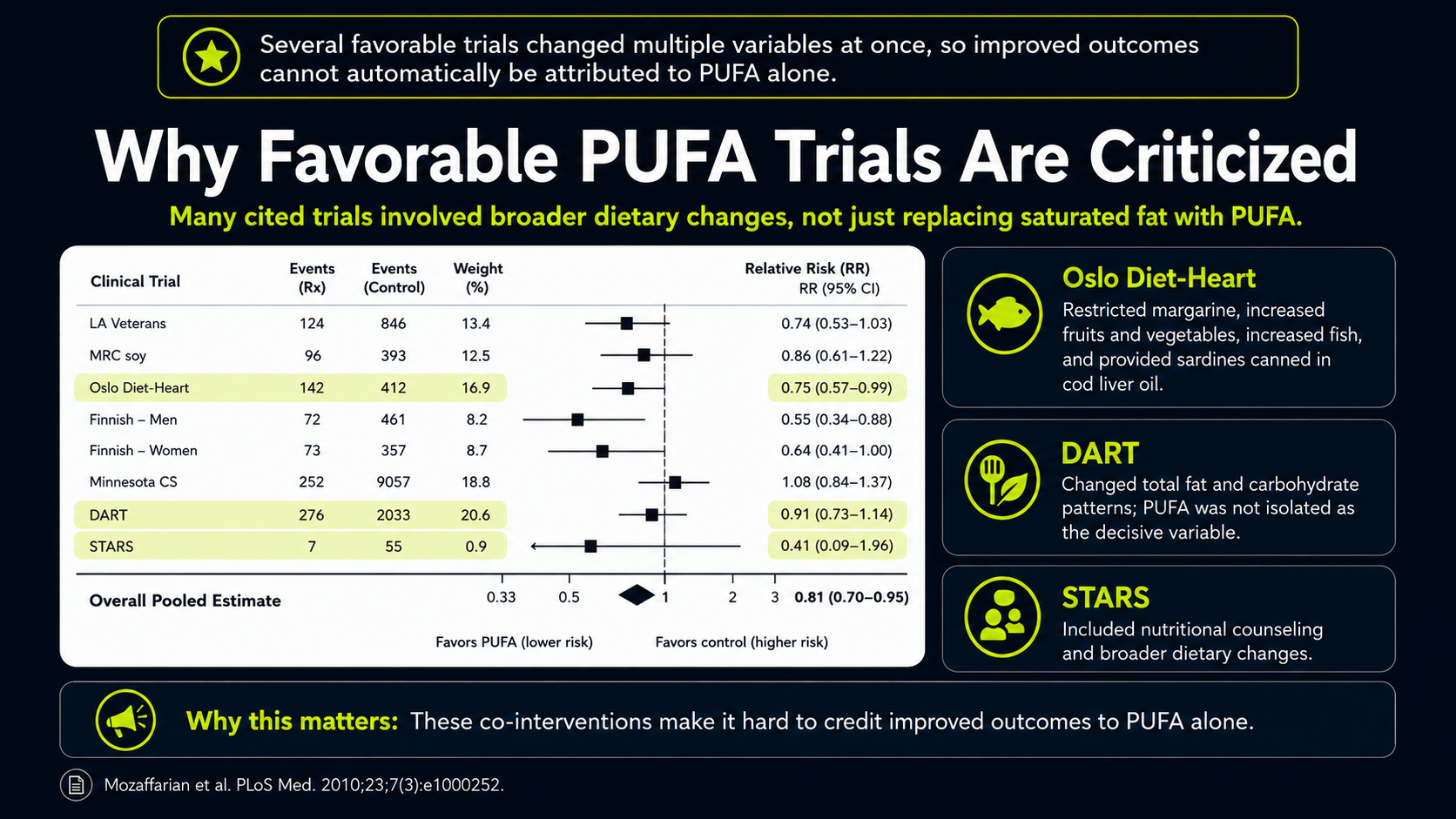
Figure 13. Trial-quality concerns in interpreting favorable PUFA-replacement evidence.
The red box and yellow callout turn the meta-analysis into a selection problem. The box highlights which trials were included, while the yellow note points out that two important trials, Rose 1965 and the Sydney Diet-Heart Study, were left out. That matters because both were unfavorable or concerning for the PUFA-replacement case. The pooled result can look more persuasive when difficult trials are excluded.
The Finnish Mental Hospitals Study raises a different problem. It is often treated as randomized evidence, but the hospital was effectively the unit of assignment, not the individual patient. One hospital received one diet while another received the comparison diet, and then the hospitals switched. That is not the same as randomizing individual people to different diets within the same setting.
This distinction is not a statistical technicality. If the hospital is the unit being assigned, then hospital-level differences--patient mix, medications, staffing, time period, and other background factors--can move with the diet. In a true individual-level randomized trial, those factors have a better chance of balancing out. Here, they remain a serious reason to avoid giving the study too much causal weight.
Several other favorable trials were not simple fat-swap experiments either. The STARS trial, or St Thomas’ Atherosclerosis Regression Study, involved more than replacing saturated fat with PUFA; it also included nutritional counseling and broader dietary changes.¹⁰
The DART trial, or Diet and Reinfarction Trial, changed total fat and carbohydrate patterns and did not cleanly isolate PUFA as the decisive variable.⁸,⁹
The Oslo Diet-Heart Study also changed many things at once: it restricted margarine, increased fruits and vegetables, increased fish, and provided sardines canned in cod liver oil.⁴,⁵
Those details matter. Fruits, vegetables, fish, sardine bones, and cod liver oil could affect vitamin C, vitamin E, glycine, minerals, fat-soluble vitamins, antioxidant defense, collagen synthesis, calcification, and inflammation. If outcomes improved, PUFA alone cannot automatically be credited.
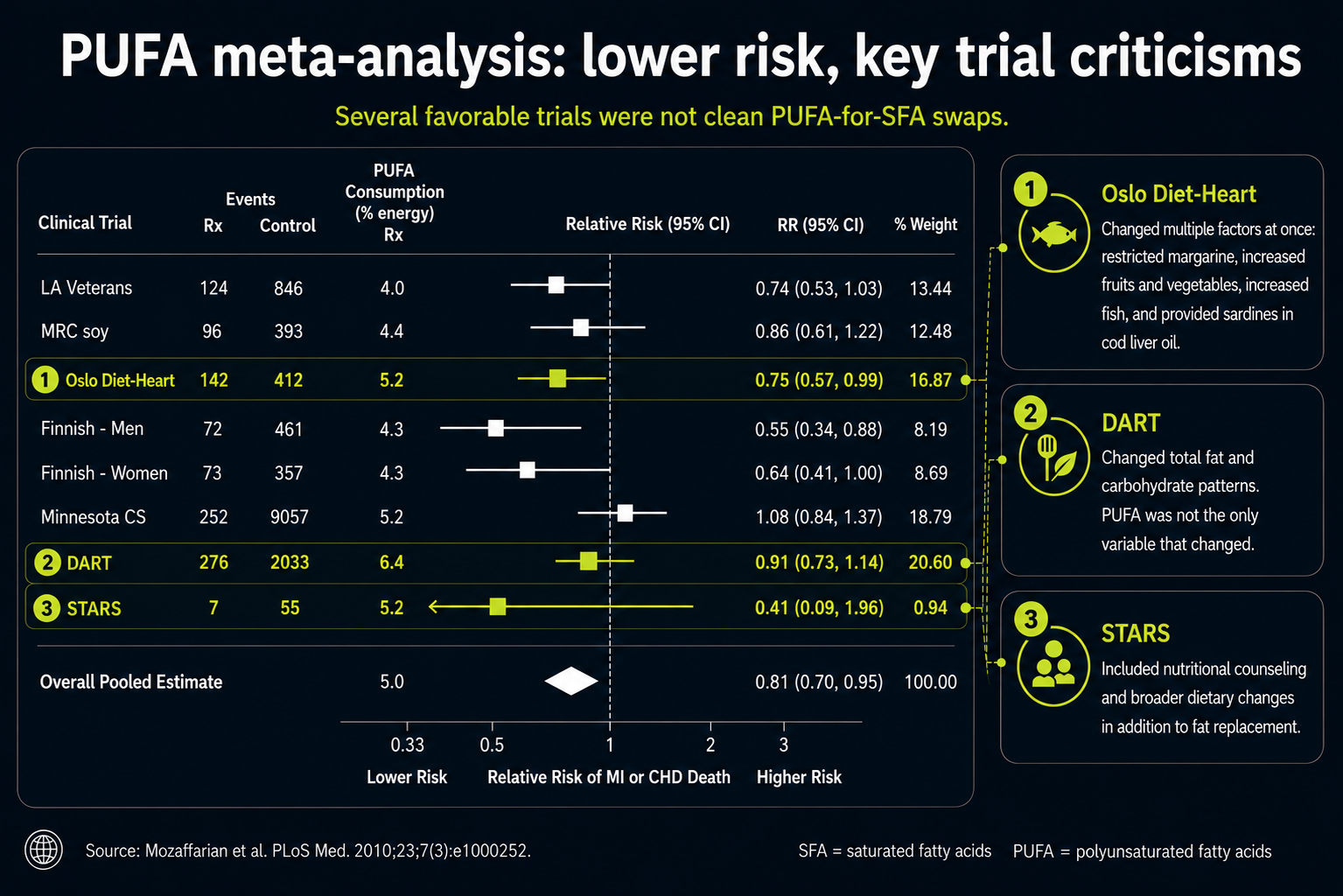
Figure 14. Mixed dietary interventions in trials that changed more than fat type alone.
The red boxes around Oslo Diet-Heart, DART, and STARS highlight a subtler issue: these trials were not clean tests of PUFA alone. They changed more than fat type, which makes them hard to use as simple evidence that replacing saturated fat with vegetable oil was the decisive factor.
That matters because several of the extra changes point in heart-protective directions for reasons unrelated to PUFA. The Oslo Diet-Heart Study, for example, included more fish, fruits, vegetables, and sardines canned in cod liver oil, while restricting margarine. Those changes could alter antioxidant intake, minerals, fat-soluble vitamins, collagen support, calcification biology, and inflammation. If outcomes improved, the improvement cannot automatically be credited to vegetable oil.
This is the larger lesson of the boxed trials: a favorable diet trial may still be telling a broader dietary-pattern story. It does not necessarily show that the PUFA-rich oil component was the protective ingredient. The distinction is central because cholesterol lowering is not the same thing as lowering oxidation, inflammation, or plaque instability.
Other trials produced mixed or concerning results. One early trial is known as the Rose corn-oil trial, published in 1965. It studied people who already had ischemic heart disease and compared usual diet with diets that added either corn oil or olive oil. The corn-oil group did not show a protective pattern under those conditions, which makes it hard to treat corn oil as a straightforward heart-protective intervention.¹
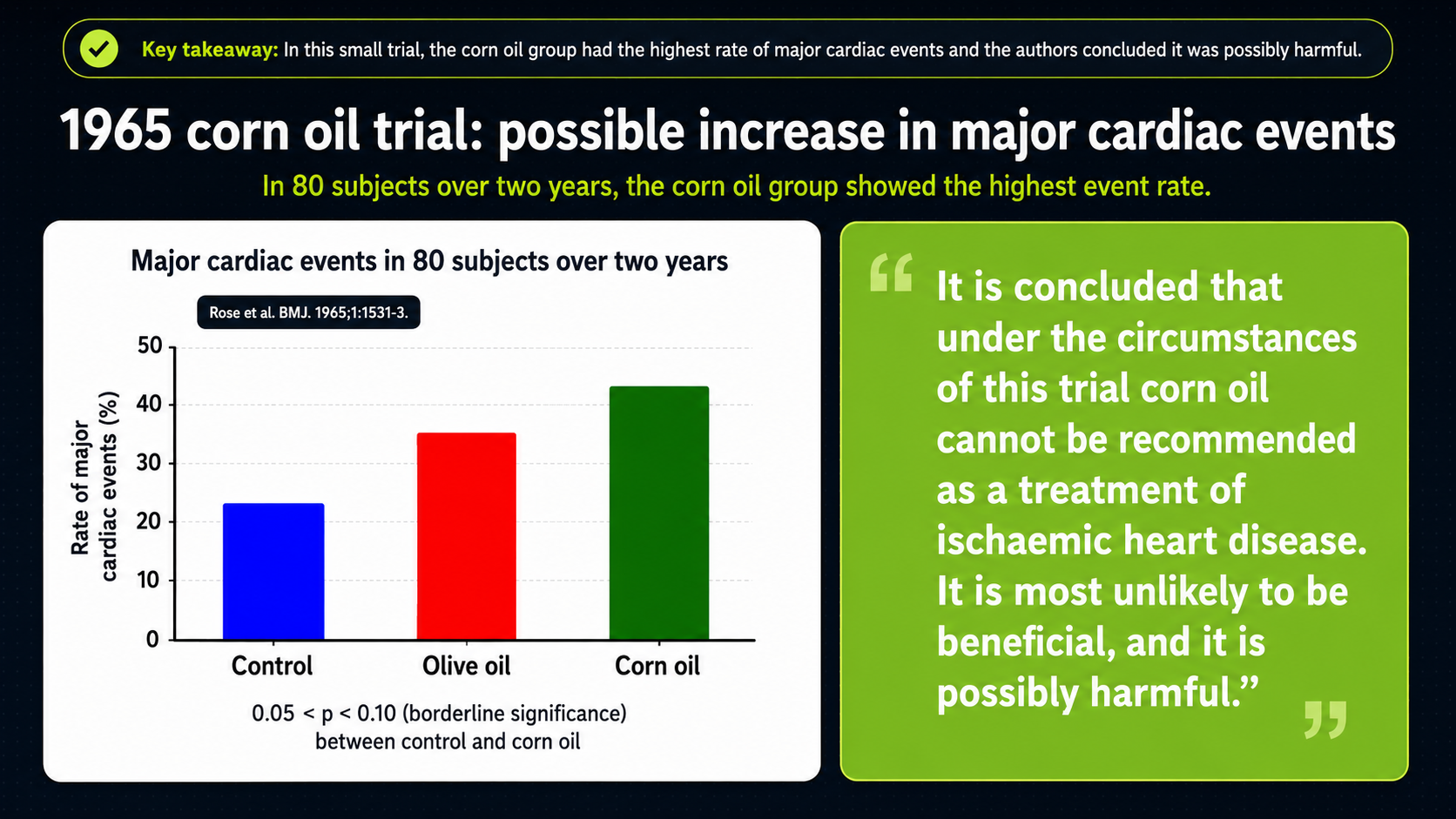
Figure 15. Rose corn-oil trial results in people with ischemic heart disease.
The Rose corn-oil trial is visually blunt. The bars compare major cardiac events in the control, olive-oil, and corn-oil groups; corn oil is the highest bar, olive oil is intermediate, and the control group is lower. In people who already had ischemic heart disease, adding corn oil did not produce the clean protective pattern the cholesterol-lowering hypothesis would predict.
That is why Rose matters even though it is an older and relatively small trial. It is one of the excluded pieces of evidence that makes the meta-analysis look less settled. If a PUFA-rich oil lowers cholesterol but does not improve hard outcomes--or appears to move them in the wrong direction--then cholesterol lowering alone cannot be treated as the whole cardiovascular story.
Another important study is the Sydney Diet-Heart Study. This trial followed men with established coronary disease and tested the replacement of saturated fat with a PUFA-rich intervention. Its results challenged the simple claim that lowering cholesterol by increasing PUFA-rich fats reliably improves survival.¹²,¹³
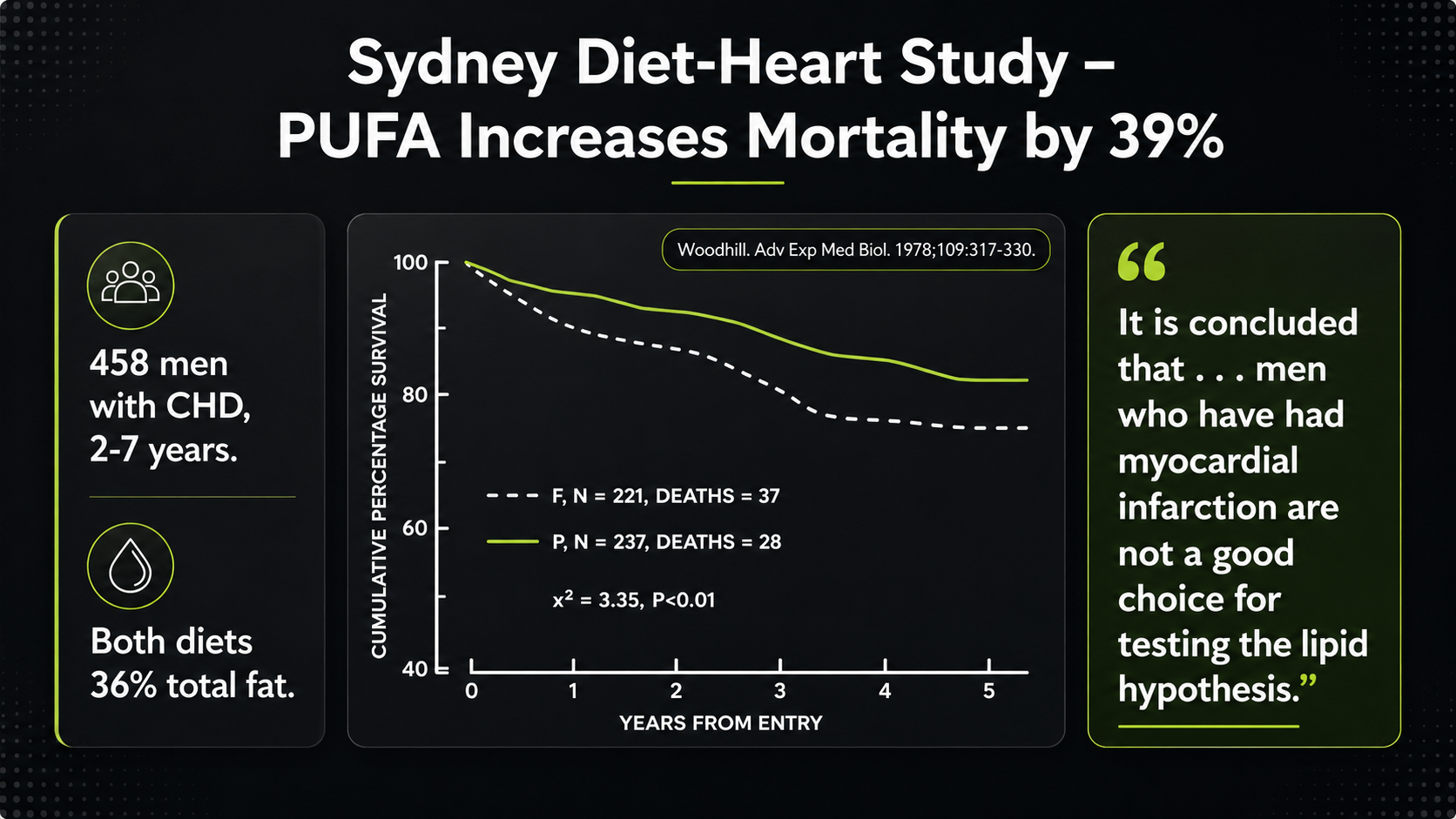
Figure 16. Sydney Diet-Heart Study survival outcomes in a PUFA-rich intervention.
The Sydney Diet-Heart Study pushes the same problem harder. The survival curve is read downward: as survival falls, more people have died. The PUFA-rich intervention line drops more than the comparison line, which is why the result is summarized as increased mortality. This is not a minor laboratory endpoint; it is the outcome people actually care about.
Sydney is especially difficult for the simple PUFA story because it involved men with established coronary disease and followed them for years. If the intervention lowers cholesterol but survival worsens, then the lipid hypothesis needs more than a cholesterol number to defend itself. The question becomes what else the diet changed inside the body--oxidation, inflammatory mediators, plaque stability, clotting tendency, or long-term vulnerability.
The Sydney source context does not add a separate dataset; it anchors the trial’s result visually. The video frame, transcript pane, and quotation all point to the same awkward conclusion: the trial’s results did not support the idea that men after myocardial infarction were a good testing ground for a cholesterol-lowering PUFA strategy.
That conclusion is revealing because it can be read two ways. One could say the population was the wrong one for the hypothesis. Or one could say the result exposed a weakness in the hypothesis itself: lowering cholesterol by increasing PUFA-rich fats may not reliably improve survival, especially in people whose arteries and heart muscle are already vulnerable.
The Minnesota Coronary Survey is also important because it was one of the stronger controlled trials in this area. It tested a cholesterol-lowering diet rich in polyunsaturated fat in institutional settings, yet it did not provide clear support for a protective effect of PUFA on hard outcomes.¹¹,¹⁴

Figure 17. Sydney Diet-Heart Study source screenshot and transcript context.
Figure 17 draws attention to the two double-blind trials in the older evidence base: LA Veterans and the Minnesota Coronary Survey. Double-blinding matters because neither the participants nor the investigators are supposed to know which group receives which diet, reducing expectation and treatment bias. In nutrition research, that level of control is rare, which is why these trials deserve special attention.
The visual point is that higher-quality evidence does not simply rescue the PUFA case. Minnesota sits close to no clear benefit, while LA Veterans supplies much of the apparently favorable signal but comes with important complications of its own. The pooled average has to be read through the anatomy of the evidence beneath it.
One of the most important trials in this debate is known as the Los Angeles Veterans Study. It began in 1959 and was published in 1969, after researchers completed an eight-year diet experiment at a Veterans Administration hospital in Los Angeles. The subjects were older male patients who lived at the facility and ate most of their meals in assigned dining halls.²,³
The study was designed to test a simple question: what would happen if a diet based more on traditional fats was replaced with one rich in polyunsaturated vegetable oils?
One dining hall served the more traditional-fat diet, including animal fats and some hydrogenated oils. The other served a similar overall menu, but replaced those fats with vegetable oils such as corn, soy, safflower, and cottonseed oil, while also limiting eggs. Color-coded meal tickets helped maintain the dining-hall assignment and blinding.
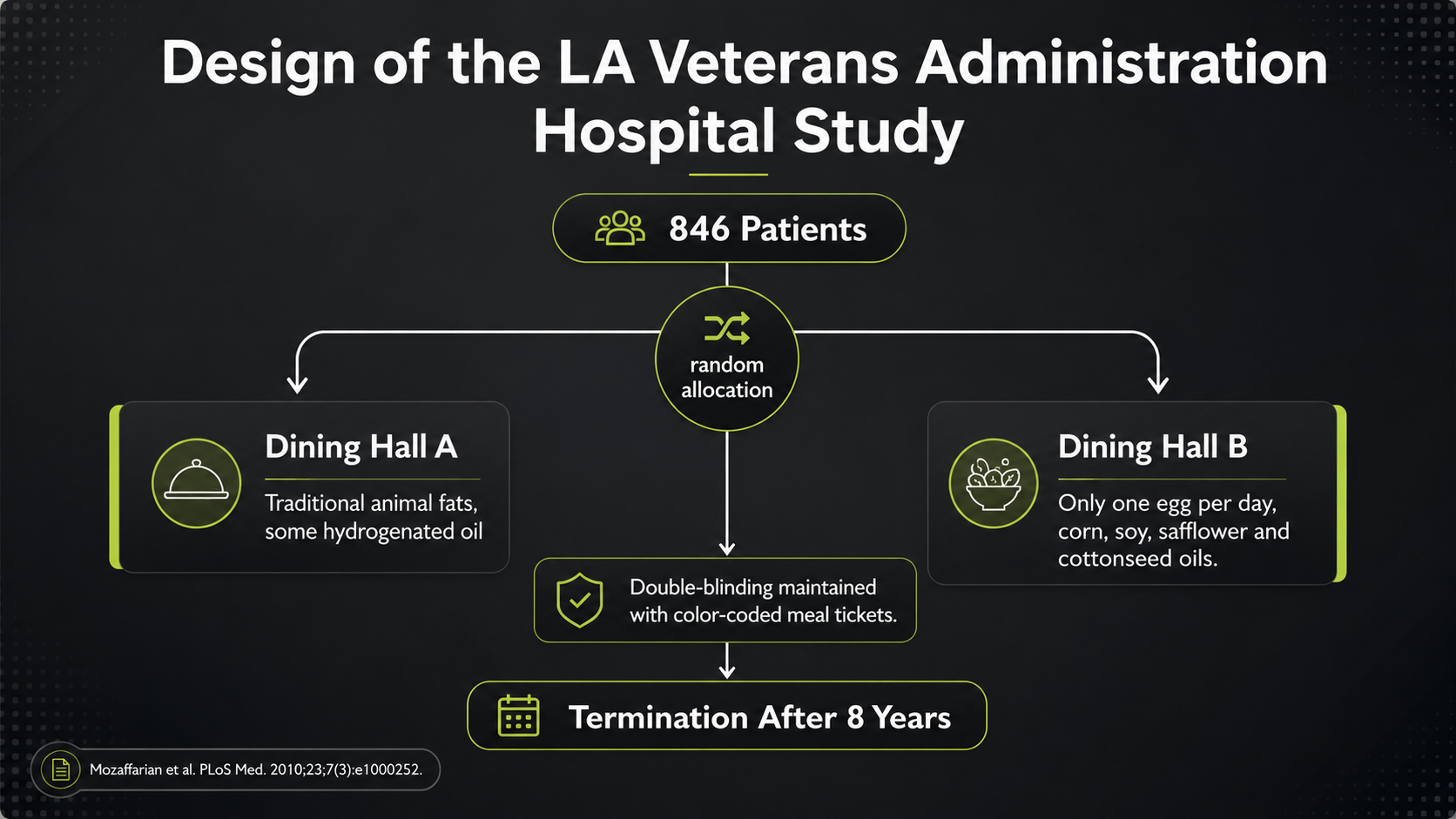
Figure 18. Los Angeles Veterans Study design, with dining-hall assignment and color-coded meal tickets.
The LA Veterans design diagram shows why the trial became so important. A total of 846 long-term Veterans Administration hospital patients were assigned to one of two dining halls. The left side represents the more traditional-fat diet; the right side represents the vegetable-oil intervention. Because meals were served in institutional dining halls and assignments were maintained with color-coded tickets, the study had far more dietary control than most free-living nutrition trials.
The intervention was meant to test a practical substitution. Dining Hall A used traditional fats, including animal fats and some hydrogenated oils. Dining Hall B served broadly similar foods but replaced those fats with corn, soy, safflower, and cottonseed oils, while limiting eggs. The design therefore asked whether a vegetable-oil-rich diet could improve long-term outcomes, not merely whether it could lower cholesterol.
That distinction is the reason this trial matters. It followed older institutionalized men for eight years and tracked deaths, not just blood numbers. If a diet looks favorable on one endpoint but unfavorable on another, the trial forces the harder question: what did it do to total mortality and long-term health?
That made the Los Angeles Veterans Study unusually important. It was not just asking whether vegetable oils could lower cholesterol. It was asking whether a vegetable-oil-rich diet improved hard outcomes over time: atherosclerotic deaths, non-cardiovascular deaths, cancer deaths, and total mortality.
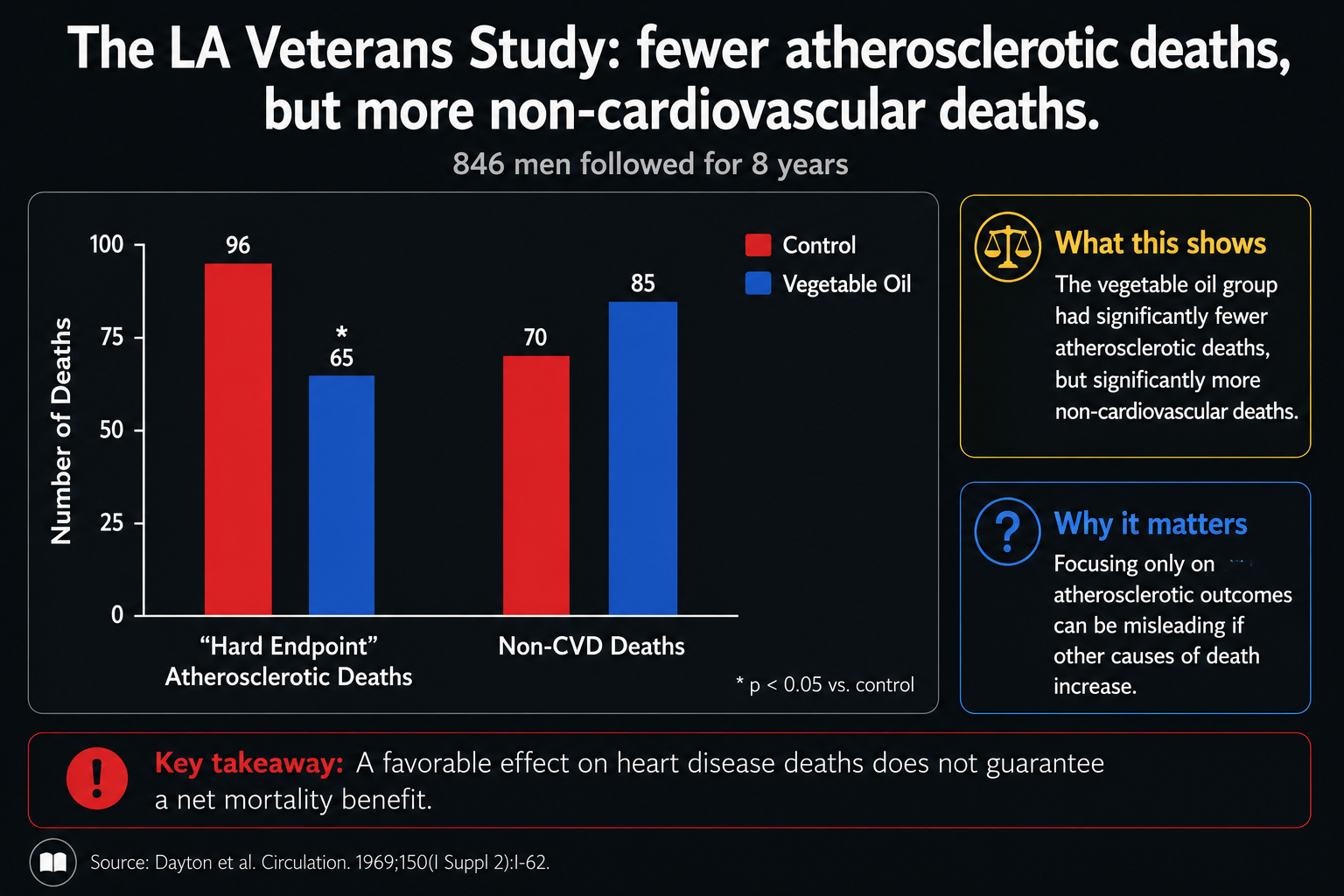
Figure 20. Hard endpoints in the Los Angeles Veterans Study, comparing atherosclerotic and non-cardiovascular deaths.
The bar chart contains the whole controversy in two pairs of columns. On the left, atherosclerotic deaths look better in the vegetable-oil group. On the right, non-cardiovascular deaths look worse. The first pair can make the intervention look protective; the second pair prevents that simple interpretation.
That is why the trial has to be judged by hard endpoints together. Fewer cardiovascular deaths would be meaningful only if they were not purchased at the cost of more deaths elsewhere. Once non-cardiovascular mortality rises, the question becomes total survival, not just whether one disease category moved in a favorable direction.
On the surface, the vegetable-oil group had fewer atherosclerotic deaths, but it had more non-cardiovascular deaths and no clear total-mortality advantage.²,³
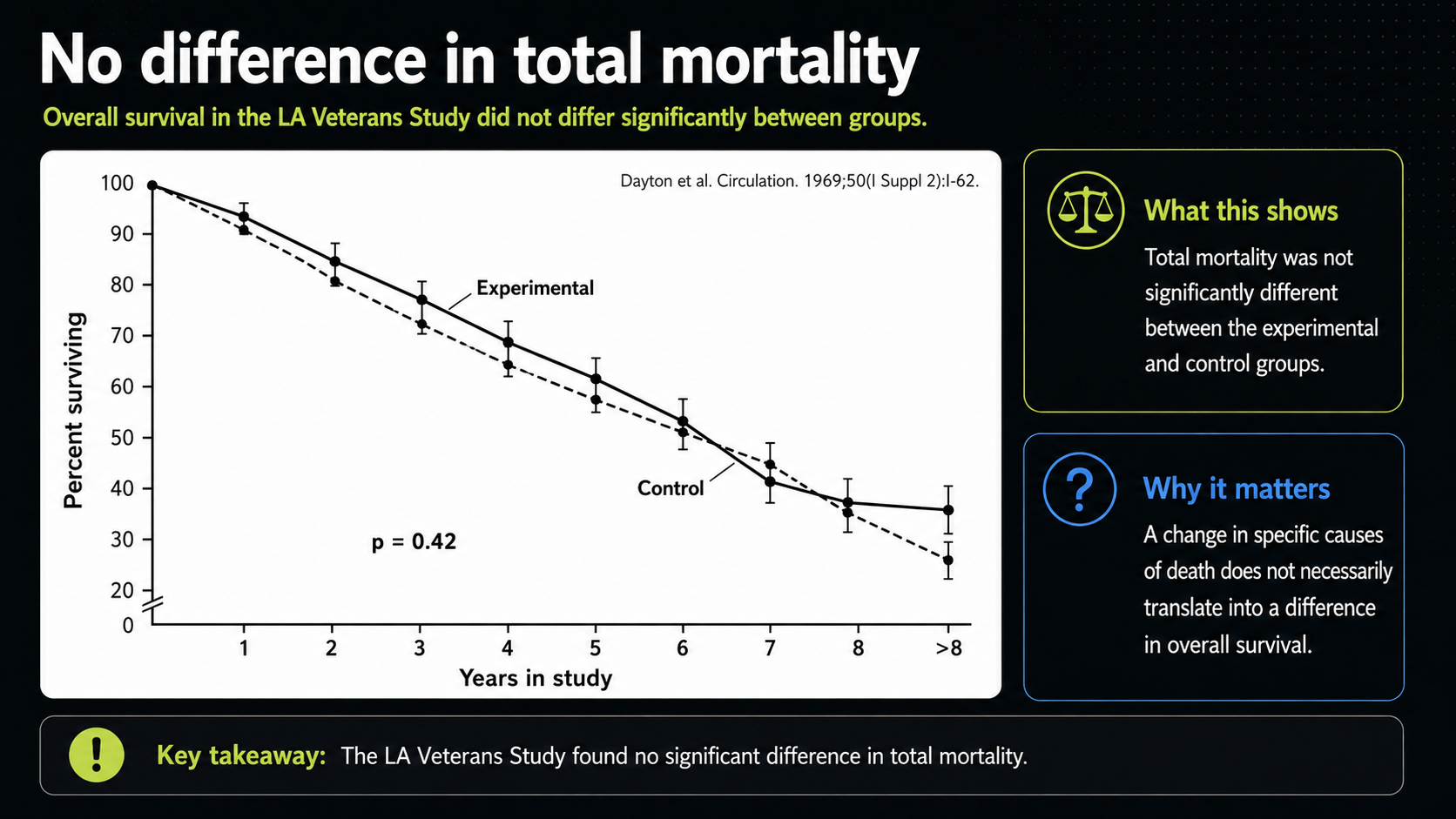
Figure 21. Total mortality in the Los Angeles Veterans Study.
The total-mortality curve gives that broader answer. The experimental and control lines cross and end close enough that the reported difference is not statistically significant. In plain terms, the vegetable-oil group did not clearly live longer. That matters because total mortality is the most unforgiving endpoint: it asks whether the intervention improved survival overall, not whether it rearranged causes of death.
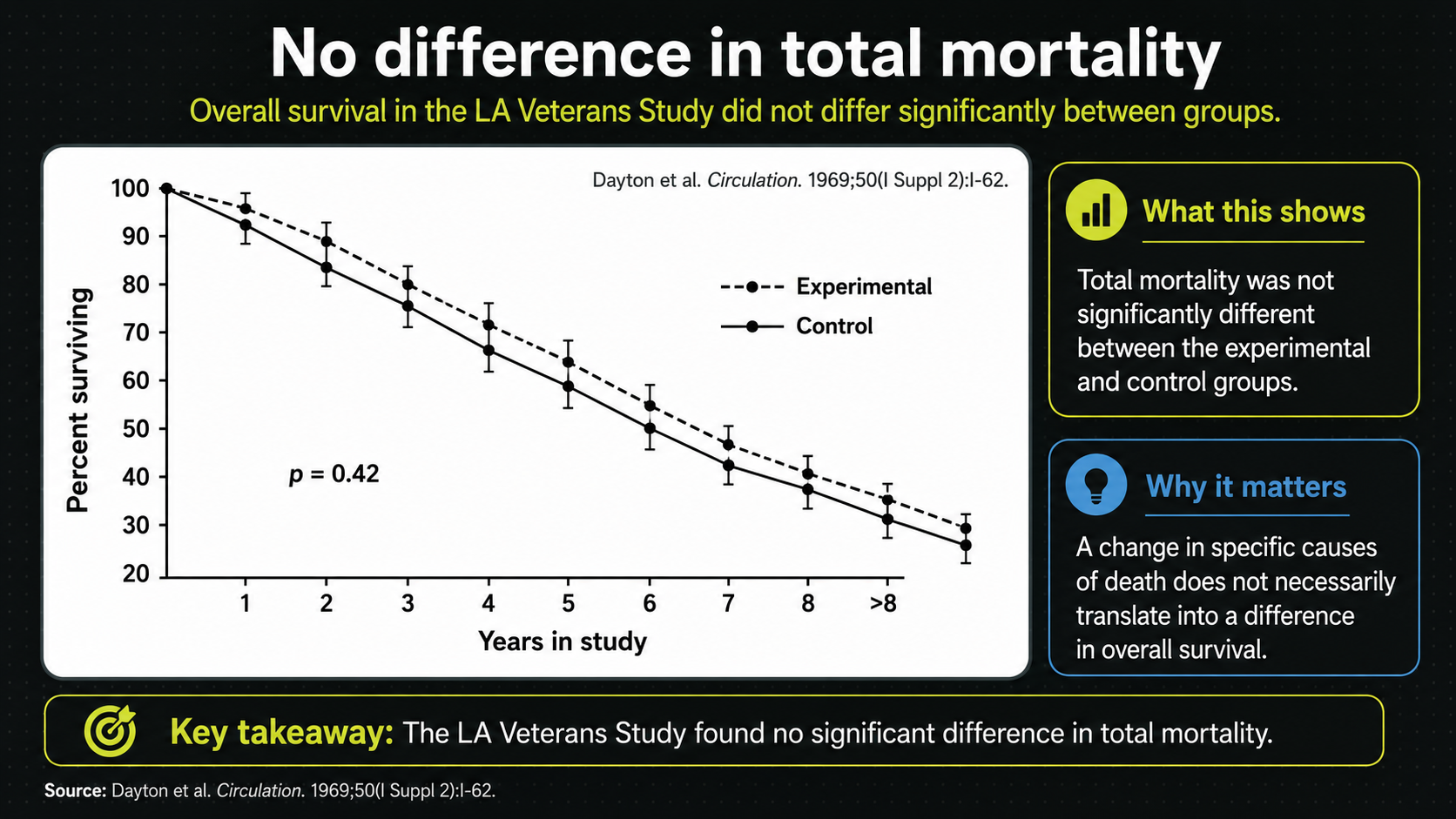
Figure 22. Total-mortality presentation of the same endpoint pattern.
This second total-mortality view reinforces the same message in the video frame. The experimental line is the PUFA/vegetable-oil group, and the control line is the traditional-fat group. Whatever the cardiovascular signal, the two survival curves do not show a clear overall advantage for the vegetable-oil diet.
The cancer and non-cardiovascular results are important because they appeared late. A shorter trial might miss them entirely. If a dietary intervention seems favorable for one endpoint but unfavorable for another, and if the unfavorable signal emerges only after several years, then short-term cholesterol lowering is a poor way to judge long-term safety.
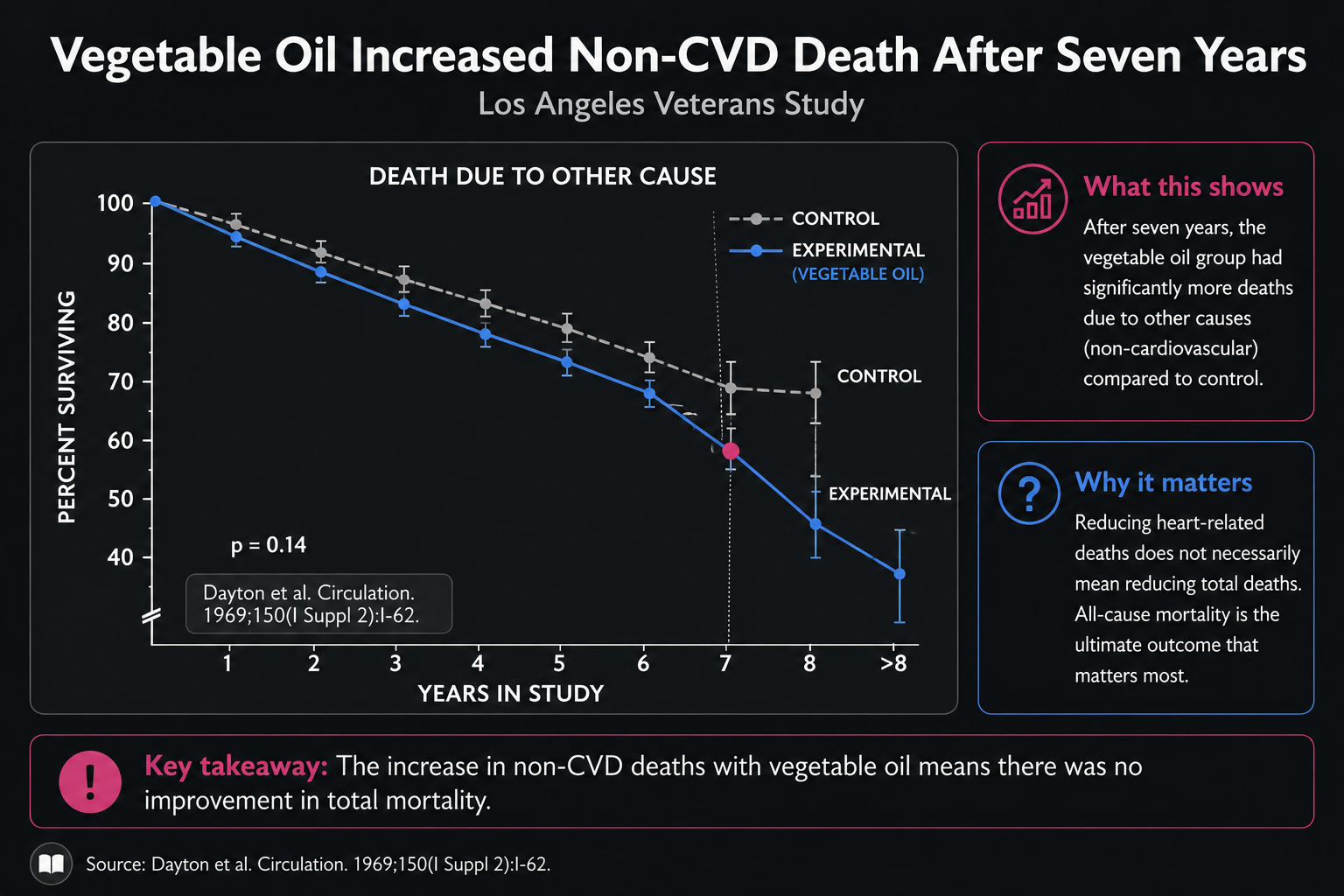
Figure 23. Non-cardiovascular deaths over time in the Los Angeles Veterans Study.
Figure 23 narrows the lens to deaths that were not cardiovascular. The timing is the important visual detail. Early on, the two lines stay close; after several years, the vegetable-oil group begins to separate unfavorably. The graph suggests that non-cardiovascular harms may take longer to become visible than cardiovascular benefits.
That timing changes how the evidence should be read. A two-year trial could miss the signal entirely. A five-year trial might only begin to see it. By seven years, the divergence becomes more obvious. If the potential harm emerges late, then short-term cholesterol lowering becomes a poor substitute for long-term outcome data.
The age of the participants sharpens the concern. As people grow older, cancer and other non-cardiovascular causes become more important relative to heart disease. If the trial had followed an older group, or had lasted nine or ten years instead of eight, the total-mortality picture might have looked different. The graph cannot prove that counterfactual, but it makes the question unavoidable.
The LA Veterans trial also shows why even favorable cardiovascular findings need careful interpretation. The control group had more smokers, including more heavy smokers, and appeared to have less vitamin E protection relative to PUFA exposure. Smoking increases oxidative stress and increases the demand for vitamin E.
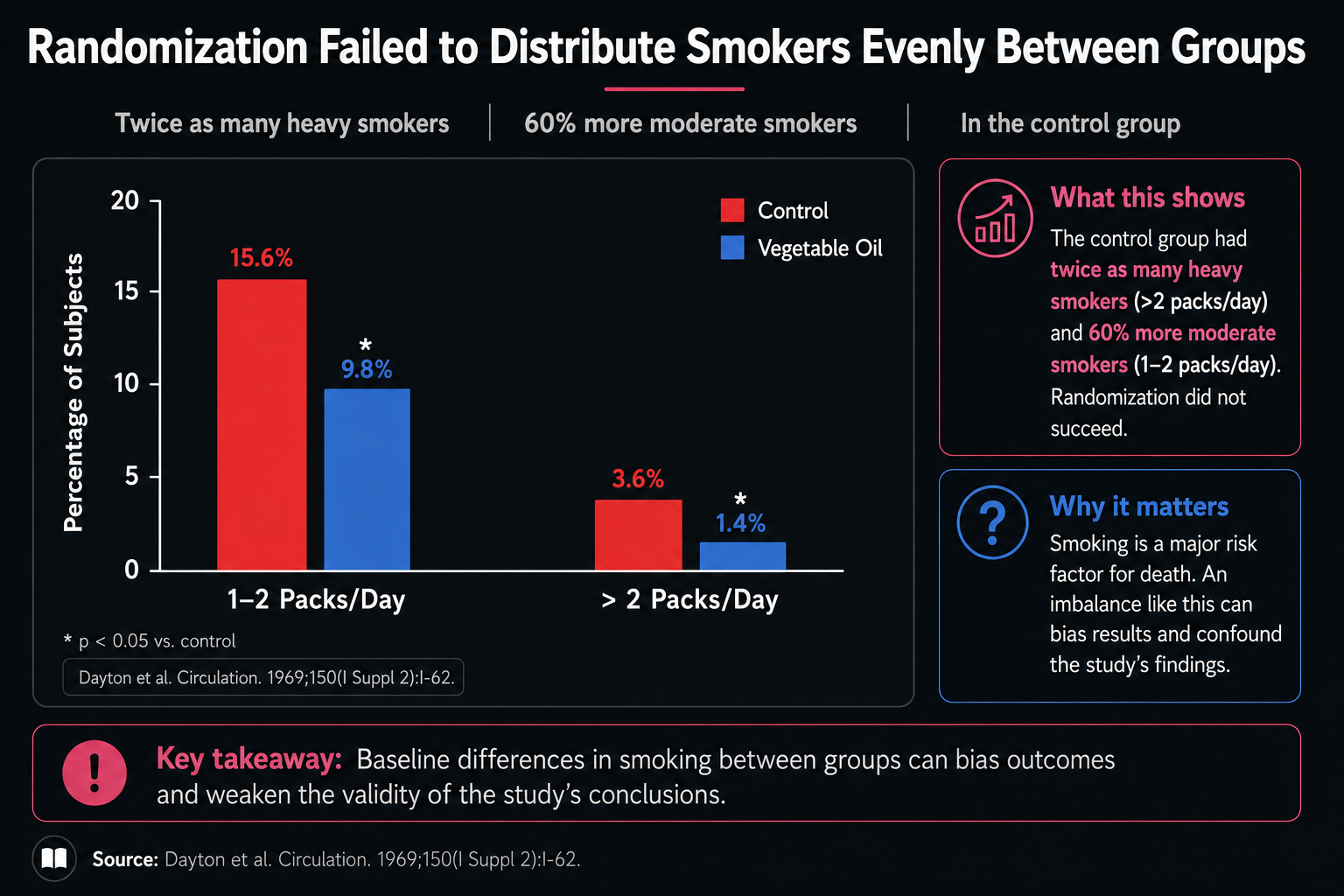
Figure 24. Smoking imbalance between study groups, complicating interpretation of the cardiovascular findings.
Figure 24 reveals a problem randomization did not solve. The red control bars are higher than the blue vegetable-oil bars for both moderate and heavy smokers, with asterisks marking notable imbalances. There were substantially more smokers in the control group, including more heavy smokers.
That imbalance matters because smoking increases oxidative stress and raises the need for antioxidant protection, especially vitamin E. If the control group had more smoking-related oxidative stress, it could have looked worse for cardiovascular deaths for reasons that were not caused by the fat intervention itself. The cardiovascular advantage of the vegetable-oil group therefore needs to be interpreted cautiously.
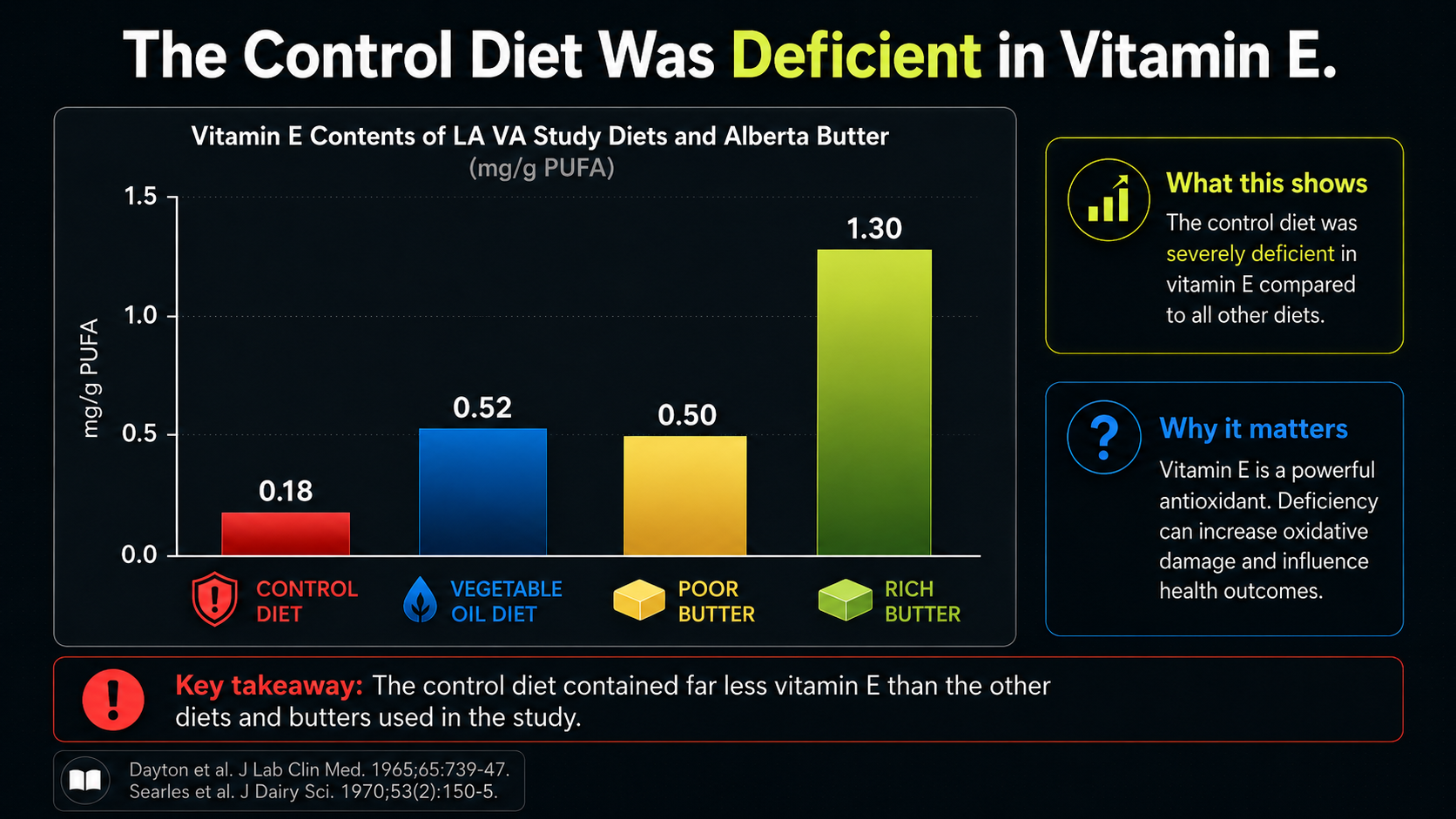
Figure 25. Vitamin E imbalance relative to PUFA exposure, adding oxidative-context concerns.
Figure 25 shifts the focus from fat type to antioxidant context. The bars normalize vitamin E to PUFA content, which is exactly the comparison that matters in an oxidation-centered biology. PUFA creates more need for antioxidant protection; vitamin E helps protect those fragile fats from peroxidation.
On that scale, the control diet looks poorly protected. It supplied much less vitamin E relative to its PUFA burden than the vegetable-oil diet did. The butter comparisons make another point: 'traditional fat' is not one uniform category. Commercial butter, poor butter, and rich spring butter can carry very different antioxidant contexts.
This does not prove that butter was the missing solution. It simply prevents a lazy comparison. A control diet low in vitamin E, combined with more smokers, could create a setting especially favorable to LDL oxidation. That would make the control arm look worse without proving that vegetable oils were broadly protective.
If excess cardiovascular deaths were concentrated among heavier smokers in the control group, then the apparent cardiovascular advantage of the vegetable-oil group may partly reflect worse oxidative conditions in the control group rather than a clean protective effect of vegetable oil.
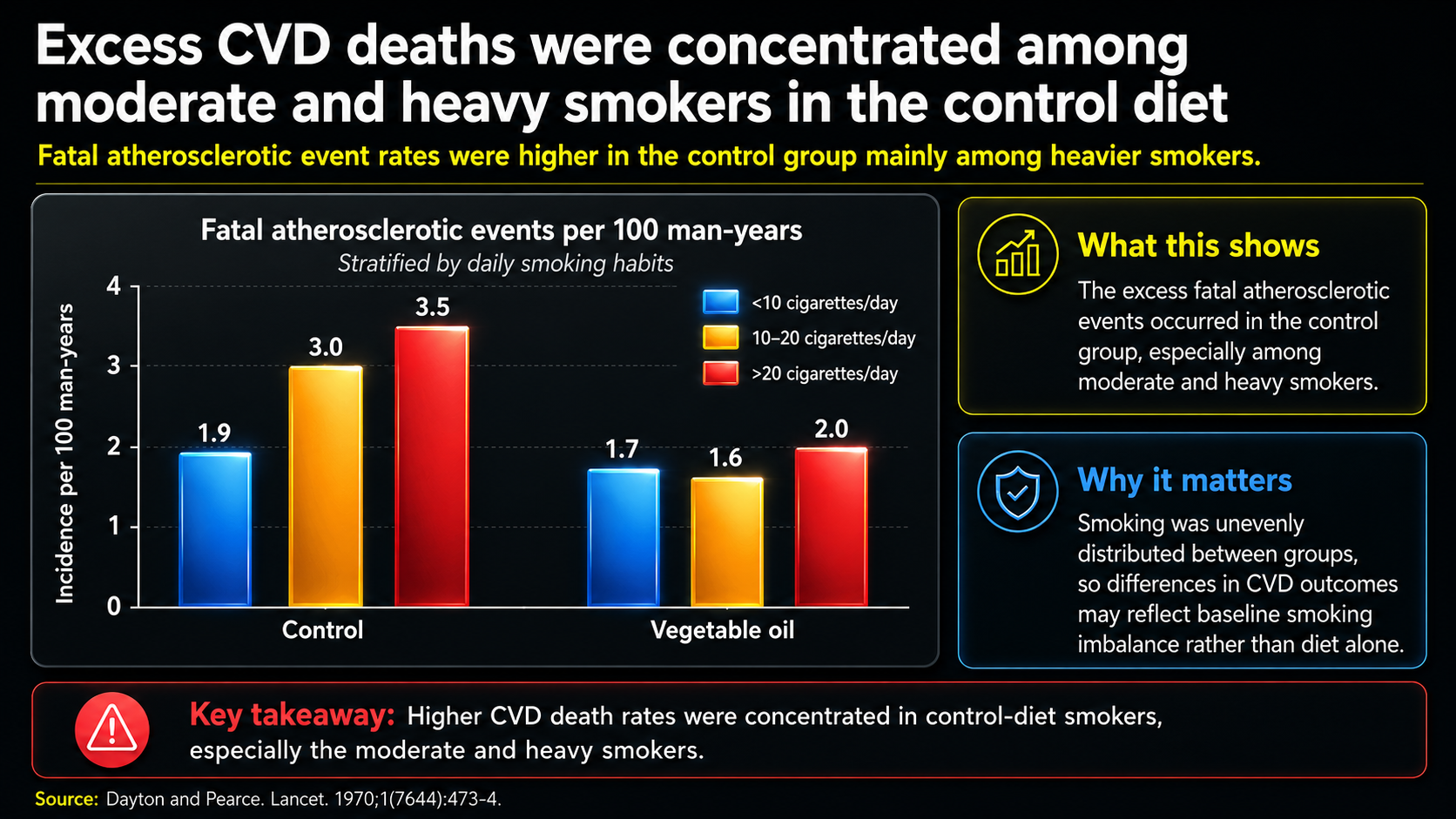
Figure 26. Excess cardiovascular deaths among moderate and heavy smokers consuming the control diet.
Figure 26 makes the smoking issue concrete. The two clusters compare fatal atherosclerotic events by smoking category within the control and vegetable-oil groups. The excess cardiovascular deaths appear concentrated among moderate and heavy smokers in the control group, not evenly spread across everyone eating the traditional-fat diet.
That pattern fits the oxidation explanation. Smoking increases vitamin E turnover, and the control diet had much less vitamin E relative to PUFA exposure. Put together, more smoking plus poorer antioxidant protection could mean more lipid peroxidation, more LDL oxidation, and more cardiovascular deaths in the control arm.
In that reading, the apparent cardiovascular benefit of the vegetable-oil diet may partly reflect a damaged comparison group. The trial still matters, but it does not cleanly prove that PUFA-rich oils are inherently protective. It shows how fat type, antioxidant status, smoking, and oxidative stress can become tangled inside a real clinical trial.
That still leaves the cancer signal unresolved. Smoking should have pushed cancer risk upward in the control group, yet the concerning cancer pattern appeared in the vegetable-oil group. That makes the trial difficult to use as simple support for PUFA-rich oils.
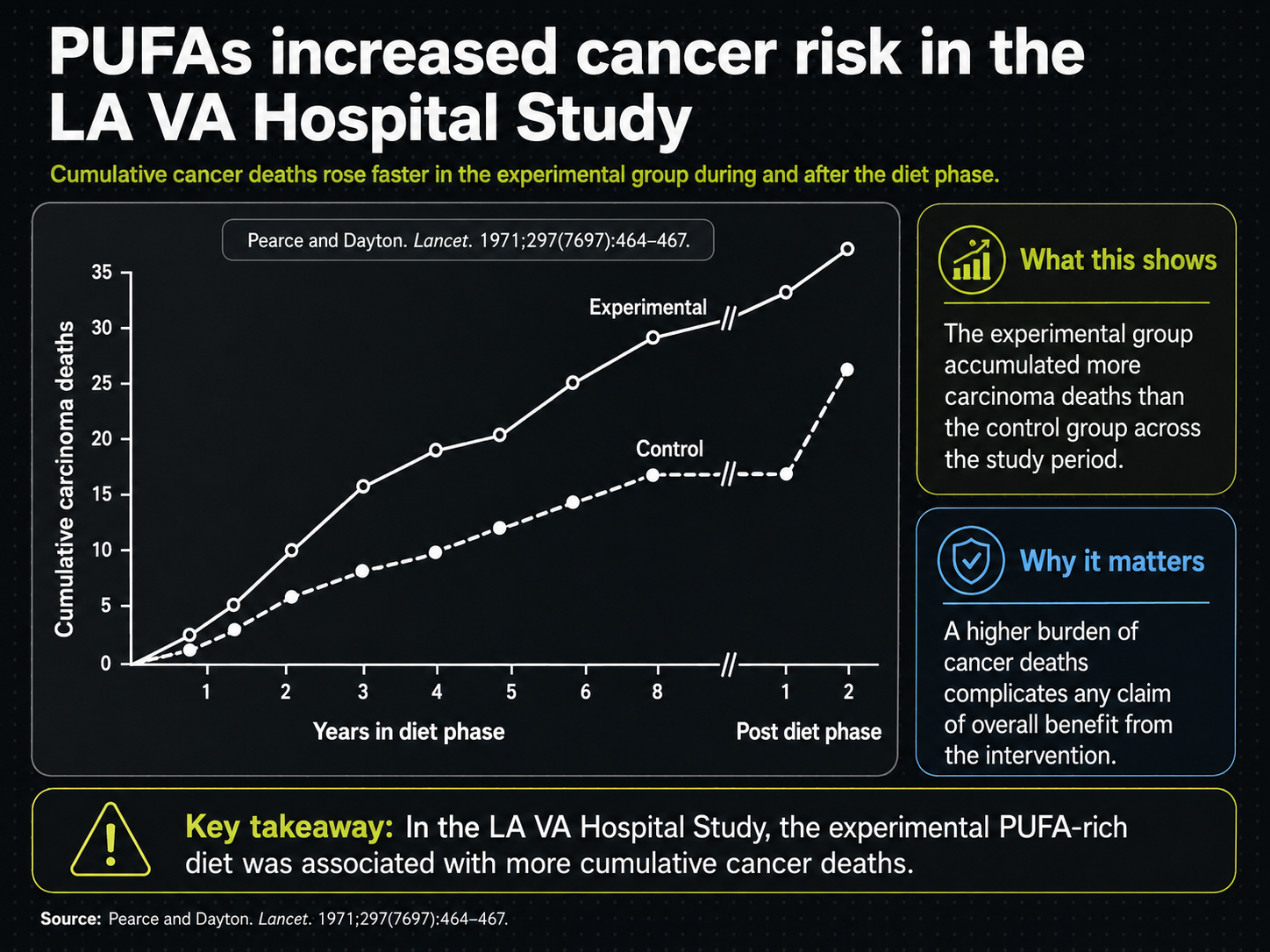
Figure 27. Cancer-risk signal in the Los Angeles Veterans Study.
Figure 27 reverses the problem. Smoking should have biased cancer risk upward in the control group, yet the experimental vegetable-oil line rises higher. The pink markings draw attention to timing: the separation begins after a couple of years and becomes more obvious after about five. That delayed pattern is why cancer and other non-cardiovascular outcomes require long follow-up.
The cancer signal is not easy to dismiss. If the group with fewer smokers still shows more cancer deaths, then smoking cannot explain the unfavorable direction. The trial does not settle every question about causation, but it makes PUFA-rich oils hard to defend on cholesterol lowering alone.
The delay also matters for the evidence base as a whole. Most diet trials were five years or shorter. If a major adverse signal requires more than two years to appear and more than five years to become clear, then many trials were simply too short to see the full tradeoff.
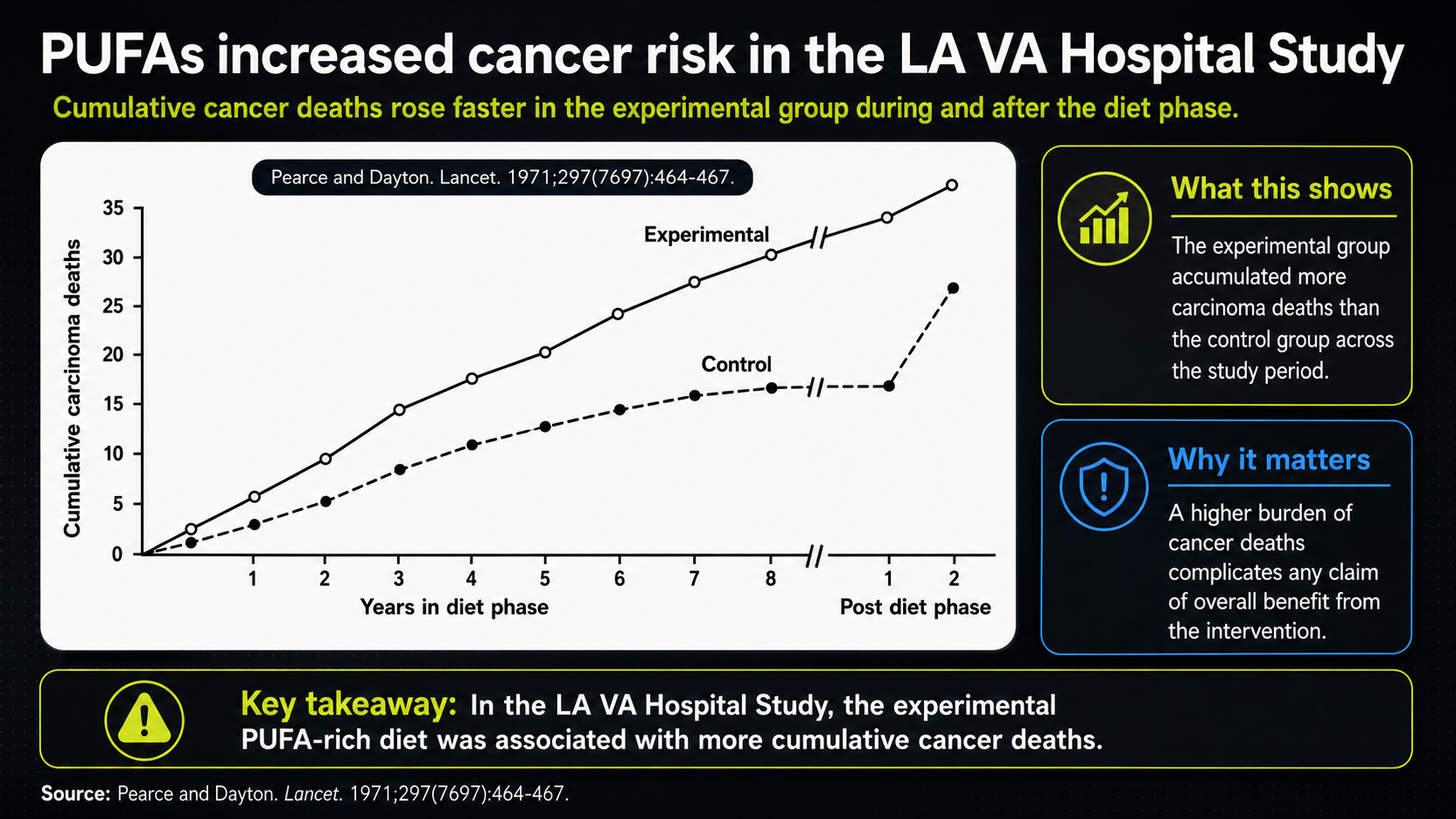
Figure 28. PUFA and cancer-risk source context for the unresolved cancer signal.
This source context preserves the cancer-risk claim without creating a new dataset. It shows the same cancer curve inside the video with the transcript pane visible, documenting where the delayed cancer-risk signal entered the lecture.
The unsettling point is that the cancer analysis was not stratified the way the cardiovascular analysis was. One would expect more cancer in heavier smokers, and the control group had more smokers. Yet the higher cancer pattern appeared in the vegetable-oil group. That is why the LA Veterans Study raises a lingering question about whether longer trials would have shown the non-cardiovascular signal more strongly.
The point is not that every study says the same thing. The point is that the evidence does not justify judging vegetable oils by cholesterol lowering alone. If the heart-disease process depends on oxidation, inflammation, immune activity, plaque stability, and clotting, then diet has to be evaluated by those processes too.
A food or oil that lowers cholesterol is not automatically protective. It must also be judged by whether it lowers the body’s burden of oxidation-prone PUFA, reduces lipid peroxidation, limits inflammatory mediator production, supports antioxidant defense, maintains tissue repair, improves overall survival, and protects long-term health.
A more coherent strategy would be to improve LDL clearance and reduce LDL oxidation without increasing the oxidation-prone PUFA content of LDL membranes. That points toward limiting PUFA exposure, supporting thyroid function, improving insulin status, controlling inflammation, strengthening antioxidant defense, and improving tissue repair rather than relying on vegetable oils as the central solution.
The dietary evidence shows the limits of judging heart disease through cholesterol alone. PUFA may lower cholesterol while still supplying the oxidative and inflammatory chemistry that makes heart disease more dangerous. But atherosclerosis is only one side of heart disease. The other side is the heart muscle itself.
The LA Veterans authors recognized this problem themselves. They noted that the small excess of non-atherosclerotic mortality late in the study raised the question of whether future trials of diets rich in unsaturated fat would need to last well beyond eight years rather than the usual five. The implication is sobering: the trials long enough to answer that question were never really done.
This is the larger warning about meta-analyses. When the underlying studies are numerous, similar, and high quality, pooling them can clarify the big picture. But when the studies are few, heterogeneous, differently designed, and sometimes confounded or selectively included, averaging them can blur the very details that matter most. A pooled diamond can look cleaner than the evidence underneath it.
A more coherent strategy would be to improve LDL clearance and reduce LDL oxidation without increasing the PUFA content of LDL membranes. That points toward thyroid function, insulin status, inflammation control, antioxidant defense, and tissue repair rather than relying on vegetable oils as the central cardiovascular solution. The practical question is not simply how to lower cholesterol. It is how to make the whole lipoprotein-and-artery system less oxidizing, less inflamed, and better able to repair.
Heart Disease Is Also a Disease of Energy
Atherosclerosis explains the disease process inside arteries, but it does not explain every form of heart disease. The next layer is the heart muscle itself. The heart can also fail when it cannot produce enough energy to contract, relax, regulate minerals, and maintain its structure.
Cardiovascular disease often refers to the blood-vessel side: atherosclerosis, calcification, narrowing, plaque rupture, clotting, heart attack, and stroke. Heart failure refers more directly to the weakening or malfunction of the heart muscle.
These two categories can reinforce each other, but they are not identical. A narrow artery can injure the heart muscle, and a weak heart can worsen circulation, kidney function, blood pressure regulation, swelling, and tissue oxygenation.
Heart failure occurs when the heart cannot pump properly, relax properly, or both. It is often described structurally, but at the cellular level it is deeply energetic.
The heart is a muscle that requires continuous ATP production. Energy is needed not only for contraction but also for relaxation. Relaxation requires the cell to move calcium back out of the contractile machinery. Energy is also required to regulate sodium, potassium, magnesium, water, and cellular structure.
When a heart cell lacks energy, it cannot maintain these gradients properly. Calcium can accumulate. Sodium and water regulation can become disturbed. The cell can swell. A swollen, calcium-loaded muscle cell cannot contract or relax normally.
This is why heart failure should be understood partly as a bioenergetic disease. The heart does not fail only because arteries are narrowed. It can fail because its cells cannot produce enough energy to maintain normal function.
Stress Chemistry Can Weaken the Heart
Chronic stress changes the chemical environment of the heart and blood vessels. Adrenaline, cortisol, aldosterone, estrogen signaling, free fatty acids, inflammatory mediators, and mineral shifts can all affect cardiovascular function.
These systems may be useful in acute stress, but chronic activation can become harmful. They also determine when stored PUFA is released from tissues and pushed into circulation.
This is where PUFA, estrogenic stress, vitamin E, clotting, and fibrosis begin to overlap. PUFA supplies the raw material for inflammatory lipid mediators. Estrogenic stress can push the body toward leakier vessels, higher inflammatory signaling, and more fibrotic repair.
Vitamin E sits on the opposite side of that equation by helping protect fragile fats from oxidation. The issue is not any one molecule in isolation, but the internal climate that decides whether repair stays orderly or turns into swelling, clotting, scarring, and stiffness.
Cortisol is catabolic. Over time, it can contribute to tissue breakdown. Adrenaline mobilizes fuel, but chronically it can increase free fatty acid release and metabolic strain. Aldosterone helps retain sodium, but it can also promote potassium and magnesium loss. Those minerals are important for cellular relaxation and electrical stability.
Estrogen signaling matters because it can interfere with both sides of heart-muscle function. The ability of the heart to contract strongly is called an inotropic effect. The ability of the heart to relax properly is called a lusitropic effect. A heart that cannot contract well cannot pump enough blood; a heart that cannot relax well cannot fill and reset properly for the next beat.
From a bioenergetic point of view, chronic estrogenic stress, cortisol, adrenaline, elevated free fatty acids, and mineral loss all push the heart toward the same failure pattern: lower energy availability, more calcium retention, more swelling, weaker contraction, and poorer relaxation. When the released free fatty acids are highly unsaturated, they add another problem: they are more vulnerable to oxidation and more likely to feed inflammatory signaling.
The common theme is that chronic stress chemistry can push the heart away from efficient energy production and stable regulation. The heart may become more dependent on stress fuels, more vulnerable to swelling and calcium overload, and less able to contract and relax normally.
This does not replace artery-wall biology. It completes it. Heart disease is not only about the blood vessels that feed the heart. It is also about the metabolic condition of the heart muscle itself.
Glucose Oxidation Supports Heart Function
The heart can use both fatty acids and glucose for fuel, but those fuels do not have identical effects.
Efficient glucose oxidation supports ATP production, carbon dioxide production, and stable cellular regulation. During chronic stress, high free fatty acids can interfere with glucose metabolism and reduce energetic efficiency.
Stress hormones increase lipolysis, releasing free fatty acids into the blood. If the stored fat released during stress is rich in PUFA, stress does not merely mobilize fuel; it mobilizes oxidation-prone material.
If free fatty acids remain high, the heart may have more difficulty oxidizing glucose efficiently. Polyunsaturated fatty acids add another concern because they are vulnerable to oxidation and can contribute to inflammatory lipid mediators.
This is central to the bioenergetic view: excessive fatty acid oxidation can suppress glucose oxidation, while efficient glucose oxidation supports the heart’s ability to contract, relax, and maintain ion balance. The issue is not merely how much fuel is available; it is whether the heart can use fuel in a way that produces energy cleanly and maintains structure.
Free fatty acids are not just passive fuel. When stress hormones keep releasing fatty acids from stored tissue, the heart may be forced into a metabolic state that is less efficient and more inflammatory. Highly unsaturated fatty acids are especially concerning because they are more vulnerable to oxidation, can produce inflammatory mediators, and can interfere with the clean glucose oxidation the heart needs for efficient contraction and relaxation.
A heart that oxidizes glucose efficiently is better able to produce energy, regulate calcium, relax properly, and maintain structure. A heart flooded with stress fuels may have more difficulty maintaining efficient respiration. The heart is not merely a pump; it is living tissue whose mechanical function depends on cellular respiration.
Thyroid Function Connects Lipids and Energy
Thyroid hormone connects lipid handling with energy production. It supports LDL receptor activity, which helps clear LDL particles from the blood. Faster LDL clearance can reduce the time LDL spends in circulation, which may reduce opportunities for oxidation.
Thyroid hormone also supports metabolic rate and mitochondrial function. The heart needs this support because it must produce energy continuously. Poor thyroid function can therefore contribute to both sides of cardiovascular disease: poorer lipid handling and weaker heart muscle energy production.
This is why thyroid function belongs in the broader biology of heart disease. It affects cholesterol metabolism, glucose oxidation, heart rate, tissue repair, and energy production. A normal cholesterol number cannot replace healthy metabolic function.
Blood Pressure Is a Whole-Body Regulation Problem
Blood pressure, gut inflammation, clotting, hormones, minerals, and blood volume are not side issues. They shape the internal environment in which arteries and heart muscle either recover or deteriorate. By this point, the pattern is the same: the cardiovascular system is responding to the whole body, not merely to one number on a lab report.
Blood pressure is often reduced to sodium intake, but the system is more complicated. Sodium, aldosterone, blood volume, kidney function, protein status, albumin, potassium, magnesium, stress hormones, and cellular hydration all interact.
When blood volume is low or sodium is inadequate for the body’s needs, aldosterone may rise. Aldosterone helps retain sodium, but it can also increase potassium and magnesium loss.
Without enough potassium and magnesium, cells may have more difficulty relaxing. This affects both the heart and blood vessels.
Low blood volume can also cause the body to constrict blood vessels to preserve circulation. That may be useful in an emergency, but chronically it can strain the cardiovascular system.
A body with low effective blood volume may try to maintain pressure through vessel constriction, stress hormones, kidney signals, and aldosterone. That can keep circulation going in the short term while increasing cardiovascular strain over time.
Protein and liver function also matter because albumin helps maintain blood volume and carries hormones, nutrients, and fatty acids through the blood. If albumin is low, blood volume and nutrient delivery can suffer.
Albumin also matters because it carries substances to cells. If albumin is inadequate, or if it is heavily occupied by free fatty acids during stress, the delivery of thyroid hormone, nutrients, and other transported substances may be impaired.
PUFA-rich free fatty acids are especially important here because they can occupy albumin while also adding an oxidation-prone burden to the blood. This links blood volume, liver function, protein status, thyroid function, PUFA burden, and free fatty acids into one regulatory network.
Blood pressure is therefore not merely a salt problem. It is a whole-body regulation problem involving minerals, protein, liver function, kidney function, blood volume, stress hormones, albumin transport, cellular hydration, and cellular energy.
The Gut Can Drive Cardiovascular Inflammation
The digestive system can influence heart disease through inflammation.
One pathway involves endotoxin, also called lipopolysaccharide. Endotoxin is produced by bacteria. If poorly digested food reaches the colon and bacterial metabolism increases, endotoxin production may rise. If endotoxin enters the bloodstream, the immune system treats it as a threat. That can increase systemic inflammation.
Because inflammation is central to atherosclerosis, chronic endotoxin exposure may contribute to vascular disease. It can promote endothelial dysfunction, immune activation, inflammatory signaling, and plaque instability.
This also brings the liver and bile into the picture. Bile acids, made from cholesterol, help digestion and help limit the absorption of irritating bacterial products from the intestine into the bloodstream.
When bile flow is poor, when liver function is strained, or when the intestinal barrier is weakened by stress, poor nutrition, or low energy production, the gut can become a larger source of systemic inflammation. Cardiovascular health depends partly on whether the intestine and liver are keeping that inflammatory material contained.
Serotonin is another gut-linked factor. Most serotonin in the body is produced in the gut. Serotonin can influence clotting, vascular tone, fibrosis, and heart function.
Fibrosis is excessive connective tissue formation. In the heart, fibrosis can make tissue stiffer and less functional. In blood vessels, fibrosis and inflammation can interfere with normal regulation.
This makes the intestine relevant to both heart attack and stroke. If endotoxin increases inflammatory signaling, if serotonin increases clotting tendency or fibrosis, and if the blood becomes more inflammatory and thrombotic, then the gut can influence whether a vulnerable plaque remains contained or becomes part of an acute event.
The gut is therefore not separate from cardiovascular disease. Digestion, bacterial metabolism, endotoxin, serotonin, inflammation, clotting, and fibrosis can all influence the cardiovascular environment.
The point is not that every person responds identically to every food. The point is that digestion changes the internal environment carried by the blood. Poor digestion, excessive bacterial fermentation, endotoxin exposure, and serotonergic signaling can all increase the burden placed on the vascular system.
The intestine belongs in any complete account of heart disease because the gut helps determine the inflammatory burden carried through the circulation.
Aspirin Shows That Heart Disease Is Also Inflammatory and Thrombotic
Aspirin is commonly associated with heart attack and stroke prevention because it reduces platelet aggregation. That alone shows that clotting is central to acute cardiovascular events.
But aspirin also illustrates a broader point. Its effects involve inflammation, prostaglandins, platelet activity, serotonin, and fatty acid metabolism. Prostaglandins and related inflammatory mediators are made from polyunsaturated fatty acids, which is why PUFA burden sits upstream of many inflammatory pathways.
The point is narrow but important: aspirin does not justify casual use; it shows that acute cardiovascular events involve inflammatory and thrombotic chemistry, not cholesterol alone.
Whether a plaque rupture becomes a heart attack or stroke depends partly on clotting tendency, platelet activation, inflammatory mediators, serotonin, prostaglandins, free fatty acids, and the condition of the blood itself.
A cholesterol-only explanation cannot fully account for that.
Treatment Should Not Focus Only on Isolated Markers
Modern treatment often focuses on measurable markers such as cholesterol, blood pressure, and imaging findings. These markers can be useful, but they can also narrow the explanation too much.
The problem with a marker-only view is that it can mistake the measurement for the disease. Cholesterol, blood pressure, and imaging findings may reveal something important, but they do not explain the whole process that produced the damage.
Lowering cholesterol does not automatically correct PUFA burden, oxidative stress, inflammation, thyroid function, glucose oxidation, endotoxin exposure, serotonin, collagen repair, mineral balance, or heart muscle energy production.
Reducing fluid does not automatically correct the stress hormones and mineral losses that may contribute to heart failure. Relaxing blood vessels does not automatically correct the metabolic reason those vessels are constricted.
The goal is not to ignore standard markers, but to treat them as clues that point back to the underlying process.
Treatment also has to pay attention to the quieter signs of metabolic resilience: whether the body stays warm, sleeps deeply, digests food comfortably, maintains adequate protein status and albumin, holds blood volume without excessive stress signaling, and repairs tissue without drifting into chronic inflammation or fibrosis.
These signs do not replace medical evaluation. They show why cardiovascular disease develops in a whole person, not in a lab value.
The better question is why LDL is oxidizing, how much PUFA is available to oxidize, why the artery wall is inflamed, and why the body is producing inflammatory lipid mediators.
It also asks why the fibrous cap is weak, why the blood is prone to clotting, why the heart muscle is energy-deficient, why blood pressure is elevated, and why the organism is relying on stress chemistry instead of efficient energy metabolism.
These questions lead to a more complete understanding of cardiovascular disease.
The Unified Biology of Heart Disease
Heart disease can be understood as a failure of regulation across several connected systems.
In the artery wall, the failure appears as PUFA peroxidation, LDL oxidation, endothelial injury, immune cell accumulation, foam-cell formation, necrotic core development, fibrous-cap weakening, calcification, rupture, and clotting.
In the blood, the failure appears as oxidized lipoproteins, PUFA-derived inflammatory mediators, free fatty acids, endotoxin, serotonin, platelet activation, low antioxidant protection, and stress hormones.
In the heart muscle, the failure appears as poor ATP production, calcium retention, swelling, impaired contraction, impaired relaxation, fibrosis, and structural weakening.
In blood-volume regulation, the failure appears as low effective circulation, vessel constriction, aldosterone activation, potassium and magnesium loss, kidney strain, albumin problems, and poor nutrient delivery.
In the whole organism, the failure appears as chronic stress, high PUFA exposure or tissue PUFA, poor thyroid function, impaired glucose oxidation, excessive fatty acid oxidation, low antioxidant defense, poor protein status, poor albumin production, mineral imbalance, digestive inflammation, and inadequate tissue repair.
This explains why cholesterol alone cannot explain heart disease. Cholesterol participates in the process, but it does not account for the inflammatory environment, PUFA peroxidation, oxidative damage, immune response, plaque instability, clotting tendency, heart muscle failure, blood-volume stress, gut-derived inflammation, or systemic metabolic strain.
Heart disease is not one isolated defect. It is a breakdown in the body’s ability to maintain vascular structure, blood stability, tissue repair, blood volume, mineral balance, and cellular energy. PUFA is not the whole disease, but it is the central dietary substrate that can feed and amplify many of the oxidative, inflammatory, and stress-driven failures.
Practical Implications
The practical implication is not to chase one marker in isolation. A coherent prevention or recovery framework would ask whether each intervention lowers oxidative pressure, reduces PUFA peroxidation, improves LDL clearance, stabilizes plaque structure, supports glucose oxidation, maintains minerals and blood volume, protects collagen repair, and reduces the inflammatory load coming from stress and digestion.
In dietary terms, the framework favors lowering long-term exposure to easily oxidized vegetable-oil PUFA, maintaining adequate protein and collagen-supporting nutrients, protecting fats with antioxidant systems, supporting thyroid and glucose metabolism, and avoiding patterns that chronically raise free fatty acids and stress hormones.
In medical terms, none of this means ignoring cholesterol, blood pressure, imaging, medication, or acute care. It means those tools should be interpreted inside the larger biological question: what is damaging the artery wall, what is weakening the plaque cap, what is making the blood more thrombotic, and what is depriving the heart muscle of energy?
Conclusion
The familiar picture of heart disease is too small. Heart disease is not a simple problem of clogged arteries. Arteries are living organs, and plaque forms inside the artery wall through oxidation, inflammation, immune activity, and repair.
LDL becomes dangerous when the PUFA-rich parts of the particle oxidize and the damaged particle is retained in the artery wall. Oxidized LDL injures the endothelium, attracts immune cells, produces foam cells, and contributes to plaque formation.
Plaque becomes dangerous when its fibrous cap weakens. A heart attack often occurs not because an artery slowly fills like a pipe, but because an unstable plaque ruptures and triggers clot formation.
This changes how diet should be judged. Polyunsaturated fats may lower cholesterol, but because they are vulnerable to oxidation and serve as precursors for inflammatory mediators, their value cannot be measured by cholesterol lowering alone.
A dietary intervention should be judged by its effects on PUFA burden, lipid peroxidation, inflammation, plaque stability, clotting, total mortality, and long-term health.
Heart failure adds another layer. The heart is a muscle that requires continuous energy. If the heart cannot produce enough ATP, regulate calcium, maintain mineral balance, and oxidize fuel efficiently, it cannot contract or relax normally.
Chronic stress, poor thyroid function, excess free fatty acids, PUFA release from tissues, aldosterone, endotoxin, serotonin, inflammation, and weak antioxidant defense can all contribute to cardiovascular failure.
The most complete explanation is biological rather than mechanical: PUFA peroxidation, oxidation, inflammation, immune containment, metabolic stress, impaired energy production, clotting, and failed repair converge inside one system. Cholesterol belongs in that system, but it is not the system. The focus has to shift from treating the artery as a clogged pipe to asking why the internal environment makes lipids oxidize, plaques rupture, blood clot, and the heart lose the energy needed to maintain itself.
References:
1. Rose GA, Thomson WB, Williams RT. Corn oil in treatment of ischaemic heart disease. Br Med J. 1965;1(5449):1531-1533. doi:10.1136/bmj.1.5449.1531
2. Dayton S, Pearce ML, Goldman H, et al. Controlled trial of a diet high in unsaturated fat for prevention of atherosclerotic complications. Lancet. 1968;2(7577):1060-1062. doi:10.1016/S0140-6736(68)91531-6
3. Dayton S, Pearce ML, Hashimoto S, et al. A controlled clinical trial of a diet high in unsaturated fat in preventing complications of atherosclerosis. Circulation. 1969;40(1 suppl 2):II-1-II-63. doi:10.1161/01.CIR.40.1S2.II-1
4. Leren P. The effect of plasma-cholesterol-lowering diet in male survivors of myocardial infarction. A controlled clinical trial. Bull N Y Acad Med. 1968;44(8):1012-1020.
5. Leren P. The Oslo diet-heart study. Eleven-year report. Circulation. 1970;42(5):935-942. doi:10.1161/01.CIR.42.5.935
6. Miettinen M, Turpeinen O, Karvonen MJ, Elosuo R, Paavilainen E. Effect of cholesterol-lowering diet on mortality from coronary heart-disease and other causes. A twelve-year clinical trial in men and women. Lancet. 1972;2(7782):835-838. doi:10.1016/S0140-6736(72)92208-8
7. Turpeinen O, Karvonen MJ, Pekkarinen M, Miettinen M, Elosuo R, Paavilainen E. Dietary prevention of coronary heart disease: the Finnish Mental Hospital Study. Int J Epidemiol. 1979;8(2):99-118. doi:10.1093/ije/8.2.99
8. Burr ML, Fehily AM, Rogers S, Welsby E, King S, Sandham S. Diet and reinfarction trial (DART): design, recruitment, and compliance. Eur Heart J. 1989;10(6):558-567. doi:10.1093/oxfordjournals.eurheartj.a059528
9. Burr ML, Fehily AM, Gilbert JF, et al. Effects of changes in fat, fish, and fibre intakes on death and myocardial reinfarction: diet and reinfarction trial (DART). Lancet. 1989;2(8666):757-761. doi:10.1016/S0140-6736(89)90828-3
10. Watts GF, Lewis B, Brunt JNH, et al. Effects on coronary artery disease of lipid-lowering diet, or diet plus cholestyramine, in the St Thomas' Atherosclerosis Regression Study (STARS). Lancet. 1992;339(8793):563-569. doi:10.1016/0140-6736(92)90863-X
11. Frantz ID Jr, Dawson EA, Ashman PL, et al. Test of effect of lipid lowering by diet on cardiovascular risk. The Minnesota Coronary Survey. Arteriosclerosis. 1989;9(1):129-135. doi:10.1161/01.ATV.9.1.129
12. Woodhill JM, Palmer AJ, Leelarthaepin B, McGilchrist C, Blacket RB. Low fat, low cholesterol diet in secondary prevention of coronary heart disease. Adv Exp Med Biol. 1978;109:317-330. doi:10.1007/978-1-4684-0967-3_18
13. Ramsden CE, Zamora D, Leelarthaepin B, et al. Use of dietary linoleic acid for secondary prevention of coronary heart disease and death: evaluation of recovered data from the Sydney Diet Heart Study and updated meta-analysis. BMJ. 2013;346:e8707. doi:10.1136/bmj.e8707
14. Ramsden CE, Zamora D, Majchrzak-Hong S, et al. Re-evaluation of the traditional diet-heart hypothesis: analysis of recovered data from Minnesota Coronary Experiment (1968-73). BMJ. 2016;353:i1246. doi:10.1136/bmj.i1246
15. Mozaffarian D, Micha R, Wallace S. Effects on coronary heart disease of increasing polyunsaturated fat in place of saturated fat: a systematic review and meta-analysis of randomized controlled trials. PLoS Med. 2010;7(3):e1000252. doi:10.1371/journal.pmed.1000252
16. Ramsden CE, Hibbeln JR, Majchrzak SF, Davis JM. n-6 fatty acid-specific and mixed polyunsaturate dietary interventions have different effects on CHD risk: a meta-analysis of randomised controlled trials. Br J Nutr. 2010;104(11):1586-1600. doi:10.1017/S0007114510004010
17. Farvid MS, Ding M, Pan A, et al. Dietary linoleic acid and risk of coronary heart disease: a systematic review and meta-analysis of prospective cohort studies. Circulation. 2014;130(18):1568-1578. doi:10.1161/CIRCULATIONAHA.114.010236
18. Steinbrecher UP, Parthasarathy S, Leake DS, Witztum JL, Steinberg D. Modification of low density lipoprotein by endothelial cells involves lipid peroxidation and degradation of low density lipoprotein phospholipids. Proc Natl Acad Sci U S A. 1984;81(12):3883-3887. doi:10.1073/pnas.81.12.3883
19. Quinn MT, Parthasarathy S, Fong LG, Steinberg D. Oxidatively modified low density lipoproteins: a potential role in recruitment and retention of monocyte/macrophages during atherogenesis. Proc Natl Acad Sci U S A. 1987;84(9):2995-2998. doi:10.1073/pnas.84.9.2995
20. Berliner JA, Territo MC, Sevanian A, et al. Minimally modified low density lipoprotein stimulates monocyte endothelial interactions. J Clin Invest. 1990;85(4):1260-1266. doi:10.1172/JCI114562
21. Ylä-Herttuala S, Palinski W, Rosenfeld ME, et al. Evidence for the presence of oxidatively modified low density lipoprotein in atherosclerotic lesions of rabbit and man. J Clin Invest. 1989;84(4):1086-1095. doi:10.1172/JCI114271
22. Glagov S, Weisenberg E, Zarins CK, Stankunavicius R, Kolettis GJ. Compensatory enlargement of human atherosclerotic coronary arteries. N Engl J Med. 1987;316(22):1371-1375. doi:10.1056/NEJM198705283162204
23. Mann J, Davies MJ. Mechanisms of progression in native coronary artery disease: role of healed plaque disruption. Heart. 1999;82(3):265-268. doi:10.1136/hrt.82.3.265
24. Burke AP, Kolodgie FD, Farb A, et al. Healed plaque ruptures and sudden coronary death: evidence that subclinical rupture has a role in plaque progression. Circulation. 2001;103(7):934-940. doi:10.1161/01.CIR.103.7.934
25. Skålén K, Gustafsson M, Rydberg EK, et al. Subendothelial retention of atherogenic lipoproteins in early atherosclerosis. Nature. 2002;417(6890):750-754. doi:10.1038/nature00804
26. Aikawa M, Rabkin E, Okada Y, et al. Lipid lowering by diet reduces matrix metalloproteinase activity and increases collagen content of rabbit atheroma: a potential mechanism of lesion stabilization. Circulation. 1998;97(24):2433-2444. doi:10.1161/01.CIR.97.24.2433
27. Price PA, Faus SA, Williamson MK. Warfarin causes rapid calcification of the elastic lamellae in rat arteries and heart valves. Arterioscler Thromb Vasc Biol. 1998;18(9):1400-1407. doi:10.1161/01.ATV.18.9.1400
28. Schurgers LJ, Spronk HMH, Soute BAM, Schiffers PM, DeMey JGR, Vermeer C. Regression of warfarin-induced medial elastocalcinosis by high intake of vitamin K in rats. Blood. 2007;109(7):2823-2831. doi:10.1182/blood-2006-07-035345
29. Enos WF, Holmes RH, Beyer J. Coronary disease among United States soldiers killed in action in Korea; preliminary report. JAMA. 1953;152(12):1090-1093. doi:10.1001/jama.1953.03690120006002
30. McNamara JJ, Molot MA, Stremple JF, Cutting RT. Coronary artery disease in combat casualties in Vietnam. JAMA. 1971;216(7):1185-1187. doi:10.1001/jama.1971.03180330061012
31. Berenson GS, Srinivasan SR, Bao W, Newman WP III, Tracy RE, Wattigney WA. Association between multiple cardiovascular risk factors and atherosclerosis in children and young adults: the Bogalusa Heart Study. N Engl J Med. 1998;338(23):1650-1656. doi:10.1056/NEJM199806043382302